|
||
|

|
陈祎 , 史诗, 梁冬丽, 王朝霞, 乔中东
, 史诗, 梁冬丽, 王朝霞, 乔中东
上海交通大学 生命科学技术学院, 上海200240
文献标识码: 1671-9964(2016)01-0016-06
文章编号: 1671-9964(2016)01-0016-06
收稿日期: 2015-04-29
网络出版日期: 2016-01-20
版权声明: 2016 上海交通大学期刊中心 版权所有
基金资助:
作者简介:
作者简介: 陈祎(1991-), 女, 硕士生, 研究方向:分子生物学, E-mail:chenyi527@hotmail.com;
通讯作者: 乔中东(1957-), 男, 博士生导师, 教授, 研究方向:环境因素对细胞发育的影响, E-mail:zdqiao@sjtu.edu.cn
展开
摘要
gx-50已被证明在阿尔茨海默症中具有神经保护作用, 然而其抗氧化机制在很大程度上仍然是未知的。为探究gx-50对在阿尔茨海默症中处于氧化应激状态的神经元细胞起到的保护作用, 本文通过测定细胞内活性氧(ROS)水平、细胞内总超氧化物歧化酶(SOD)活性和细胞分泌物丙二醛(MDA)含量检测gx-50在细胞水平的抗氧化能力。并检测了gx-50在细胞和组织水平对于JAK-STAT信号通路和caspase-3蛋白表达水平的影响。结果表明, gx-50可以使暴露于β淀粉样蛋白(Aβ)的PC12细胞分泌的ROS和MDA水平降低, 并保护胞内总SOD酶活性; gx-50可以在细胞和组织水平活化JAK-STAT信号通路, 可以降低PC12细胞内caspase-3蛋白活性亚基的相对表达水平; 而JAK2的特异性抑制剂, AG490, 逆转了gx-50的以上抗氧化保护作用。这表明gx-50可以通过活化JAK-STAT通路, 一定程度上减弱神经元所受到的氧化损伤, 对神经元起到保护作用, 因此在阿尔茨海默症预防治疗方面具有一定的应用前景。
关键词:
Abstract
A novel compound derived from Zanthoxylum, gx-50(N-[2-(3, 4-dimethoxyphenyl)ethyl]-3-phenyl-acrylamide), has been demonstrated that it has neuroprotective effects against Alzheimer’s disease(AD)by our previous study.This study focuses on the mechanism of its antioxidant properties against AD.We measured the levels of intracellular reactive oxygen species(ROS), superoxide dismutase(SOD)activity and malondialdehyde(MDA)to determine the anti-oxidative ability of gx-50 at cellular level.We also measured gx-50’s effect on JAK-STAT signaling pathway and the relative expression levels of activated caspase-3.Data showed that gx-50 reduced the levels of reactive oxygen species(ROS)and malondialdehyde(MDA), and recovered the activity of total intracellular superoxide dismutase(SOD)in neuronal PC12 cells exposed to Aβ.After gx-50 pretreatment, the levels of p-JAK2 and p-STAT3 both increased in PC12 cells, while they were down-regulated in Aβ-treated group.In present study, we also found that gx-50 reduced the relative expression level of the activated caspase-3 in PC12 cells by activating JAK-STAT signaling pathway.Results demonstrated that gx-50 reduced amyloid-beta(Aβ)induced oxidative stress in neuron-like PC12 cells by enhancing the activation of JAK-STAT signaling pathway.It might help to protect neurons in Alzheimer's disease.
Keywords:
阿尔茨海默症(AD)是一种与年龄相关的慢性神经退行性疾病, 其标志性病变包括大脑中神经元死亡, 蛋白质沉积物的积聚(细胞外老年斑)和神经元纤维缠结。虽然其发病机理还没有被完全研究透彻, 但越来越多的证据表明, 自由基形成和破坏之间的不平衡参与了阿尔茨海默症的发病机制[1]。衰老相关的自由基假说表明阿尔茨海默症患者和正常衰老的大脑所具有的一个共同关键特征是脑组织中氧化应激程度的增加。有解剖学证据表明, 在阿尔茨海默症小鼠模型和阿尔茨海默症患者的脑组织中, β淀粉样蛋白(Aβ)斑块的聚集部位会产生大量自由基攻击机体 [2]。氧化应激的这些标志性小分子与阿尔茨海默症的发病机制有着密切关联。因此, 如何利用抗氧化治疗中和高活性氧自由基小分子带来的神经元损伤, 已成为越来越重要的课题。本实验室的前期研究显示, 一种分离自花椒的天然化合物, gx-50(N-[2-(3, 4-二甲氧基苯基)乙基]-3-苯基丙烯酰胺), 可以通过“血脑屏障”进入中枢神经系统(central nerve system, CNS)发挥生物学功能, 在阿尔茨海默症小鼠模型中对于神经退行性病变有着积极的治疗效果。gx-50能有效地抑制由Aβ诱导的神经元凋亡, 并能够显著减弱β淀粉样前体蛋白(APP)转基因小鼠的空间记忆障碍, 提高其学习能力[3]。
细胞内氧化还原程度改变会导致氧化应激状态的形成, 进而导致神经元损伤。Janus激酶信号转导和转录(JAK-STAT)信号通路活化因子对于细胞内氧化还原反应有一定的影响[4]。本实验前期研究的基因芯片数据表明, Jak2和Stat3基因在Aβ处理组和gx-50与Aβ共处理组之间存在表达水平上的明显差异[3]。因此, 我们假设, gx-50可能经由影响JAK-STAT通路发挥其生物学功能。
在本研究中, 我们以交感神经系统神经元样细胞系PC12细胞作为主要研究材料, 对gx-50的生物学功能和氧化应激之间的联系进行了初步探讨。结果表明, 在PC12细胞中, gx-50具有抵抗氧化应激的积极作用。gx-50预处理可以在一定程度上活化JAK-STAT通路, 降低PC12细胞内各项氧化指标的水平, 并下调凋亡相关蛋白caspase-3的活化水平, 这可能有助于其在阿尔茨海默症患者中发挥对于神经元细胞的抗氧化保护作用。
合成Aβ42肽和gx-50购买自Sigma-Aldrich公司(美国); gx-50(分子式:C19H21NO3)用二甲基亚砜(DMSO)溶解成浓度为10 μmol/L的母液, 保存于-20 ℃。Aβ42肽溶解于双蒸水中, 使用之前在37 ℃孵育5 d, 以促进其纤维化[5]。JAK2的特异性抑制剂AG490(酪氨酸B42)购买自Selleck Chemicals(USA), 溶解于二甲亚砜(DMSO)中形成浓度为50 mmol/L储存液, 保存于-20 ℃。2', 7'-二氯荧光素二乙酸酯(DCFH-DA)从Sigma-Aldrich公司(美国)购得, 实验时根据具体需要用F12K培养基稀释至相应浓度。分析纯30% H2O2购自上海凌峰化学试剂有限公司, 实验时使用PBS缓冲液稀释成浓度为30 mmol/L的母液在4 ℃避光保存。
MDA检测试剂盒, SOD检测试剂盒购自碧云天生物制品公司(中国江苏)。
免疫组织化学EnVision检测试剂盒购自Dako公司(丹麦)。
神经元样PC12细胞在加入15%马血清(HS)和2.5%的胎牛血清(FBS)的F12K培养基中培养。培养环境为含5% CO2的37 ℃恒温培养箱。
将培养的PC12细胞接种在多聚-L-赖氨酸包被的6孔板上, 密度为5×106个/孔。培养12 h后, 将培养基换为无血清的F12K培养基, 并加药处理。
Aβ处理细胞组分为5组, 包括对照组和4个治疗组:gx-50(1 μmo/L), Aβ(10 μmol/L), gx-50+Aβ和gx-50+Aβ+AG490(10 μmol/L)。在gx-50+Aβ组中, PC12细胞先用gx-50预处理0.5 h, 再用Aβ42处理12 h以诱导氧化应激。gx-50+Aβ+AG490组的细胞先使用AG490(10 μmol/L)预处理0.5 h, 再采用与gx-50+Aβ组相同的方式进行处理。
过氧化氢氧化处理细胞组也分为5组, 包括对照组和4个治疗组:gx-50(1 μmol/L), H2O2(300 μmol/L), gx-50+H2O2和gx-50+H2O2+AG490(10 μmol/L)。H2O2处理时间为12 h, 处理方式与Aβ处理细胞组相同。
PC12细胞内ROS水平通过使用荧光染料DCFH-DA进行检测。
不同处理组细胞使用浓度为10 μmol/L的DCFH-DA孵育1 h, 并用PBS洗涤2次。使用荧光显微镜(DM2500, 徕卡, 德国)在相同曝光强度下进行拍摄。使用Image J软件分析荧光强度, 并与对照组相比较, 得到ROS相对表达水平。
PC12细胞内总超氧化物歧化酶(SOD)活性和丙二醛(MDA)含量使用碧云天有限公司(南通)生产的商业试剂盒分别进行测量。组织蛋白浓度使用BCA蛋白测定试剂盒来确定。这些实验严格根据制造商的说明进行。所有的实验独立重复3次。
测PC12细胞内总SOD酶活性时, 将细胞用预冷的磷酸盐缓冲盐水(PBS)洗涤2次, 重悬后在冰上匀浆, 并在4 ℃以1 000 r/min离心10 min, 取上清液进行检测。在MDA测定实验中, 收集不同处理组的细胞培养上清液作为实验材料。
细胞样品在冰上用RIPA裂解缓冲液解离30 min, 然后离心(12 000 r/min, 20 min, 4 ℃), 取上清液作为检测样品。蛋白质浓度利用BCA蛋白质测定试剂盒测定。采用标准方法进行Western印迹分析。兔抗p-STAT3多克隆抗体(SAB, 稀释1∶1 000), 兔抗p-JAK2多克隆抗体(SAB, 稀释1∶800)和兔抗caspase-3多克隆抗体(Proteintech, 稀释1∶2 000)作为第一抗体。兔抗GAPDH单克隆抗体(Proteintech, 稀释1∶3 000)作为内参。
将膜用含Tween-20的TBS缓冲液充分洗涤。膜使用HRP化学发光试剂盒(TIANGEN biotech, 北京), 曝光于X射线胶片。p-JAK2和p-STAT3的蛋白表达水平使用相对于GAPDH的光带强度比率来表示。
将切片60 ℃烘烤过夜, 恢复至室温后常规脱蜡入水。切片经消除内源性过氧化物酶活性、抗原修复, 0.01 mol/L TBS缓冲液冲洗后。使用0.1% Triton-100溶液在37 ℃打孔0.5 h, 用2% BSA/TBS溶液在37 ℃封闭1 h。加入兔抗p-STAT3多克隆抗体(SAB, 稀释1∶100), 4 ℃孵育过夜。TBS冲洗后, 使用DakoK5007抗鼠/兔通用HRP标记抗体, 稀释4倍, 37 ℃孵育1 h。切片经TBS冲洗、DAB显色、苏木素复染、脱水、封片后, 于显微镜下观察并拍照。每只小鼠随机选取10张切片, 在同一放大倍数, 同一曝光度下采集图像。
使用单因素方差分析(SPSS, 19.0版)进行统计分析, 所有实验均独立重复3~4次, 实验误差均以mean±SD计算。*表示P<0.05, **表示P<0.01, 认为具有统计学显著差异。
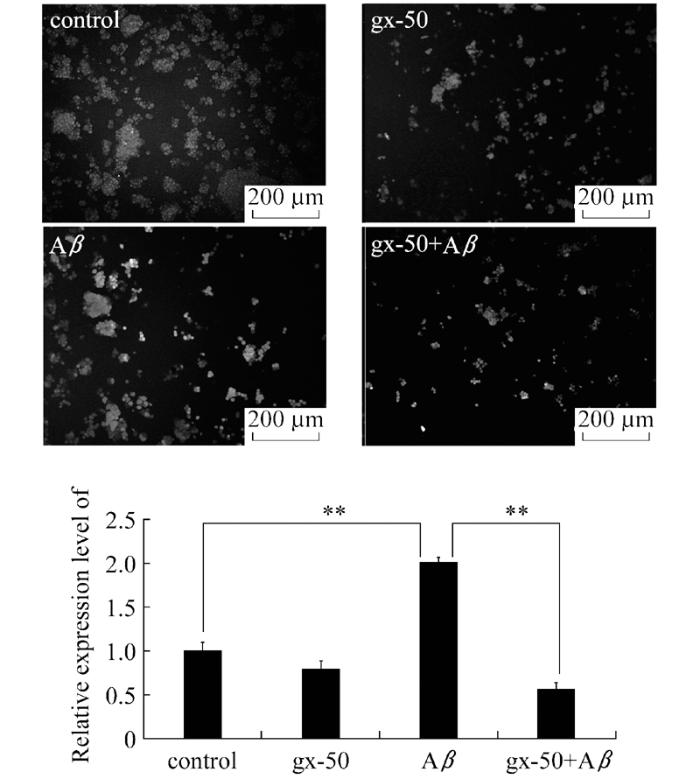
为了评估gx-50对PC12细胞氧化应激程度的直接影响, 本研究检测了PC12细胞内活性氧簇(ROS)的表达水平。通过使用Image J软件对荧光图片进行分析, 计算所得照片的平均荧光密度, 并将其与对照组相比较计算出ROS相对表达水平。统计分析后, 我们得到了更加直观的数据以显示gx-50预处理对Aβ诱导刺激的PC12细胞内ROS表达水平的影响。结果表明, 与对照组相比较, Aβ处理组中PC12细胞产生的ROS水平明显升高。使用gx-50预处理0.5 h后再加入Aβ处理, PC12细胞产生的ROS水平被下调至接近对照组水平(图1, P<0.01)。而在培养环境中没有Aβ刺激的情况下, 单独使用gx-50处理并没有明显影响PC12细胞内氧化应激程度。
以上实验结果表明gx-50可以减弱Aβ诱导的PC12细胞内ROS的过度表达, 说明了gx-50在PC12细胞内有一定的抗氧化作用, 可以减少PC12细胞在氧化应激状态下受到的自由基攻击。
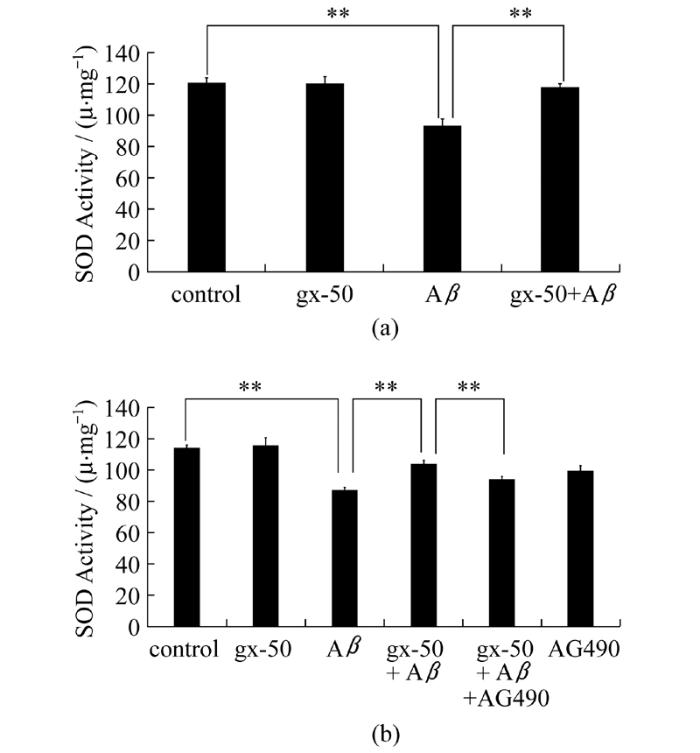
为对gx-50作用于细胞内抗氧化剂的影响进行进一步研究。本研究检测了不同处理组中细胞内总超氧化物歧化酶(SOD)的活性水平。实验数据表明, 相比于对照组中PC12细胞内的总SOD酶活性水平(97.934 U/mg), Aβ单独处理组中PC12细胞内总SOD酶活性水平显著降低(12.776 U/mg), 而gx-50显著地增强了PC12细胞内总SOD酶活性(92.417 U/mg), 减弱了Aβ对SOD的酶活性损伤(图2a, P<0.01)。
图1 gx-50对神经元样PC12细胞内ROS的表达水平的影响实验误差采用mean±SD表示, 实验重复3~4次, **表示P< 0.01
Fig.1 The effect of gx-50 on the expression of intracellular ROS in neuronal PC12 cellsThe values are expressed as mean ± SD, n=3-4.**P< 0.01
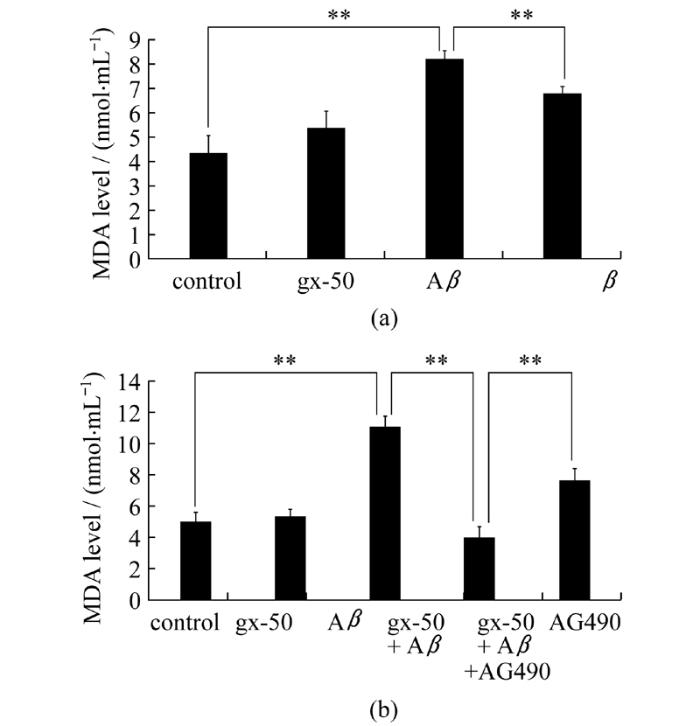
由于2.1和2.2中已经研究了gx-50对于活性氧的产生和消耗过程的影响, 于是本研究进一步选取细胞内氧化还原反应的产物作为检测对象。本研究使用微量MDA试剂盒以检测PC12细胞内的脂质过氧化产物丙二醛(MDA)的表达水平。结果显示, Aβ单独处理组中PC12细胞产生的MDA水平(8.172 nmol/mL)显著高于对照组(4.301 nmol/ mL), 而在gx-50+Aβ组有明显的降低(6.774 nmol/mL)(图3a, P<0.01)。结果表明gx-50确实在PC12细胞内对抗了Aβ导致的氧化损伤, 表现出抵抗抗脂质过氧化的功能。
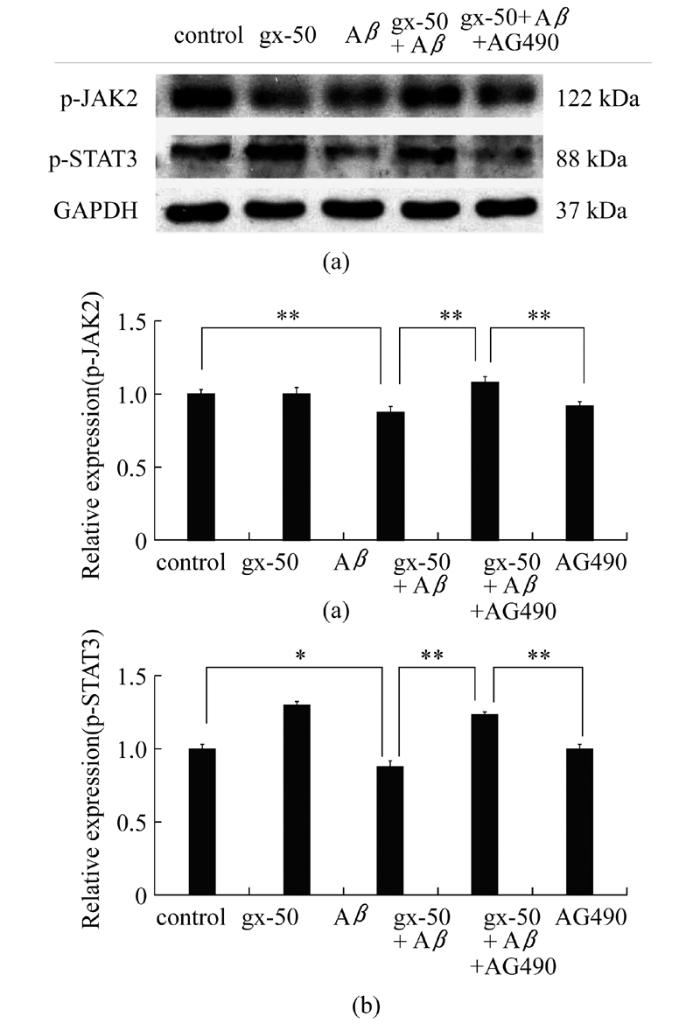
在PC12细胞内, 与Aβ单独处理组相比, gx-50+Aβ组中JAK2和STAT3的磷酸化水平显著上调(P<0.01, 图4a和4b)。而在gx-50+Aβ+AG-490组中, JAK2和STAT3的磷酸化水平Z均显著下调(P<0.01, 图4a和4b)。
免疫组织化学实验中, p-STAT3免疫阳性染色的细胞被染成棕色。免疫组化染色结果显示, 经由gx-50注射后的APP转基因阴性(APP-)小鼠的海马区域, p-STAT3免疫阳性信号明显多于使用生理盐水注射的APP-小鼠。同样的, 在APP转基因阳性(APP+)小鼠中, gx-50注射提高了海马区神经元中p-STAT3的表达量(图5)。实验结果表明gx-50治疗在APP转基因小鼠海马区域的神经元中激活了JAK-STAT通路。
图2 gx-50对神经元样PC12细胞内总超氧化物歧化酶活性的影响严格按照试剂盒说明检测各分组中PC12细胞内总SOD酶活性。实验误差采用mean±SD表示, 实验重复3~4次, *表示P<0.05, **表示P<0.01
Fig.2 Effect of gx-50 on activity of total intracellular SOD in neuronal PC12 cellsSuperoxide dismutase Assay Kits were used to measure the levels of activity of total intracellular SOD in PC12 cells after different treatments.The values are expressed as the mean±SD of three independent experiments.*P<0.05, **P<0.01
以上实验结果表明了gx-50确实在PC12细胞中和转基因小鼠海马区神经元中都对于JAK-STAT通路有一定的激活作用。
为了证实gx-50的抗氧化作用是否通过JAK-STAT通路来进行的, 我们又在增加了gx-50+Aβ+AG490组后, 对于上文的氧化指标进行检测, 探讨JAK-STAT通路的抑制剂AG490会对gx-50的抗氧化作用产生何种影响。氧化应激指标检测结果显示, 加入JAK2的特异性抑制剂AG490后, gx-50+Aβ组中本已显著减少的MDA水平(3.976 nmol/mL)又显著性地回升(7.593 nmol/mL)(图3b, P<0.01)。而AG490的加入, 使gx-50对于PC12细胞内SOD的保护作用也受到了显著抑制(图2b, P<0.05)。
图3 gx-50对神经元样PC12细胞分泌的MDA水平的影响使用微量MDA检测试剂盒检测细胞培养上清液中PC2细胞分泌的MDA水平。实验误差采用mean ± SD表示, **表示P<0.01
Fig.3 Effect of gx-50 on MDA production in neuronal PC12 cellsMDA Assay Kits were used to measure the levels of MDA in the medium of PC12 cells.The values are expressed as the mean ± SD of three independent experiments.**P<0.01
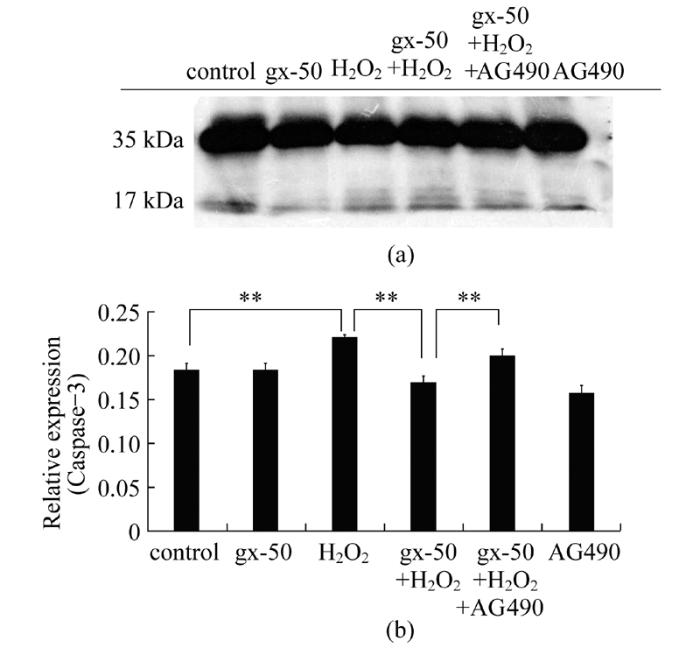
为分析gx-50对于处于氧化应激状态下的神经元细胞的保护作用, 本研究利用蛋白质印迹分析(Western blot)的手段检测了在不同分组处理下, 剪切后的活性caspase-3蛋白片段(17 kDa)相对于没有催化活性的procaspase-3蛋白(35 kDa)的表达量。与对照组比较, H2O2处理组中caspase-3蛋白17 kDa活性亚基的相对表达量有明显的升高。经gx-50预处理0.5 h后再加入H2O2处理与直接加入H2O2比较, PC12细胞内活化的caspase-3蛋白的相对表达量显著下调(图6, P<0.01)。而在gx-50+H2O2+AG490组中, PC12细胞内活化的caspase-3蛋白的相对表达量较gx-50+H2O2组显著上调(图6, P<0.01)。结果表明gx-50可以降低氧化环境下PC12细胞内的caspase-3活性亚基相对表达水平, 在一定程度上抑制氧化损伤引发的神经元细胞凋亡。并且gx-50的这一保护作用受到JAK2蛋白的特异性抑制剂AG490的负调控, 说明了JAK-STAT通路可以在一定程度上影响氧化应激状态下, gx-50对于神经元的抗凋亡保护作用。
图4 gx-50在神经元样PC12细胞内对JAK-STAT信号通路的影响采用Western blot检测PC12细胞内p-JAK2和p-STAT3蛋白的表达量(A), 其相对表达水平使用内参GAPDH蛋白表达量作参考(B)。实验误差采用mean±SD表示, 实验重复3~4次, *表示P< 0.05, **表示P< 0.01
Fig.4 Effects of gx-50 on JAK-STAT signal pathway in neuronal PC12 cellsThe expression levels of p-JAK2 and p-STAT3 were detected by Western blot(A), and their relative expressions compared with GAPDH were analyzed(B).The values are expressed as the mean ± SD of 3-4 independent experiments.*P< 0.05, **P< 0.01
大量文献报道指出, 自由基介导的氧化性损伤是许多衰老问题, 包括阿尔茨海默症的形成原因[10]。细胞内氧化水平的升高, 能够影响一些与衰老相关疾病密切关联的细胞内生物学进程[6]。活性氧增加对细胞造成的损害包括了攻击生物大分子如蛋白质, 脂质和DNA的活性[7]。MDA是细胞内脂质过氧化的直接产物, 而氧化应激不仅可导致神经元细胞分泌的MDA水平升高, 同时也损害了细胞内抗氧化酶的活性, 包括过氧化氢酶(CAT), 超氧化物歧化酶(SOD), 谷胱甘肽过氧化物酶(GPx)等[8]。表明氧化应激与衰老密切相关的遗传证据已经在不同物种的动物模型上得到了证实[9]。氧化应激在阿尔茨海默病中的重要作用已得到公认。本实验的前期研究发现gx-50可有效延缓衰老, 然而其抗氧化机制在很大程度上仍处是未知的, 因此研究gx-50针对神经元的抗氧化保护功能对理解其在阿尔茨海默症中的抗氧化作用具有重要的意义。
图5 免疫组织化学实验检测gx-50在APP-Tg小鼠脑组织内对JAK-STAT信号通路的影响p-STAT3免疫阳性的细胞被染成棕色。细胞核使用苏木素复染
Fig.5 Effect of gx-50 on JAK-STAT signaling pathway in the brain tissues of APP-Tg mice observed with immunohistochemical stainingThe p-STAT3 positive neurons was stained dark brown by DAB.The nucleus were stained blue by hematoxylin
图6 gx-50在PC12细胞内对活化的caspase-3蛋白相对表达水平的影响采用Western blot检测PC12细胞内caspase-3蛋白的表达量(A), 其活化水平用caspase-3蛋白的17 kDa亚基相较于35 kDa的procasepase-3蛋白的相对表达量表示(B)。实验误差采用mean±SD表示, 实验重复3~4次, **表示P<0.01
Fig.6 Effects of gx-50 on the relative expression levels of active casepase-3 in PC12 cellsThe expression levels of active casepase-3 were detected by Western blot(A), and their relative expressions compared with procaspase-3 were analyzed(B).Data were mean ± SD error; n=3-4.** P<0.01
Aβ是阿尔茨海默症主要形成原因, 促进了阿尔茨海默症形成和发展过程中自由基和氧化损伤的产生[1]。本研究使用Aβ处理神经元样PC12细胞, 并发现Aβ确实导致了神经元细胞氧化应激状态的形成。
本实验室的前期实验已经表明, gx-50在体外可以使Aβ寡聚体解聚[12]。已有明确的实验证据证实, gx-50无论是对原代培养的新生大鼠脑神经元细胞还是在APP-Tg小鼠模型中都具有强大的神经保护能力[12]。而且在Aβ存在的环境下, gx-50预处理可以抑制小胶质细胞的过度趋化性迁移[13]。本研究的结果显示, gx-50减少了细胞内ROS和MDA的水平, 并保护了在Aβ诱导的氧化应激的条件下神经元样PC12细胞内总SOD酶活性。实验数据证实gx-50对处于Aβ诱导的氧化应激状态下的神经元细胞具有一定的抗氧化保护作用。
Aβ导致的JAK2-STAT3通路在海马神经元的年龄依赖性失活与阿尔茨海默症相关的记忆损伤联系密切[14]。已有研究表明该通路中的STAT3蛋白在阿尔茨海默症患者的海马神经元中活性降低[15]。在STAT3基因敲除的转基因小鼠中, 神经系统中保护性蛋白质会减少[16]。
因此, 本研究提出gx-50可能作为JAK-STAT通路的活化剂, 以减弱阿尔茨海默症中神经元细胞遭受的氧化损害的假说。为了进一步探索、证实这一假说, 本研究检测了不同分组处理下, PC12细胞中JAK2和STAT3的磷酸化水平变化。
本研究的实验结果显示, gx-50可显著提高神经元样PC12细胞内p-JAK2和p-STAT3的表达水平。并通过检测PC12细胞内氧化应激指标, 发现gx-50显著的减少了由Aβ诱导的氧化应激状态下PC12细胞内ROS和细胞外MDA的过量表达, 并有效地保护了PC12细胞内总SOD酶活性。而gx-50的抗氧化保护功能, 可以被JAK2的特异性抑制剂AG 490显著地逆转。这表明gx-50确实通过激活JAK-STAT通路实现其抗氧化功能。
本实验室的前期研究表明, gx-50能够抑制Aβ诱导的神经元凋亡, 并影响凋亡相关基因的表达[12]。为了进一步探究在氧化应激环境下, JAK-STAT通路是否影响gx-50对于神经元的抗凋亡保护作用, 本研究检测了在H2O2诱导的氧化应激环境下, 不同处理组PC12细胞内caspase-3蛋白的17 kDa活性亚基相对表达水平的变化。
caspase-3是参与细胞凋亡级联反应的重要激酶, 在神经细胞中表达较高, 是检测神经细胞凋亡的重要指示蛋白之一。无催化活性的procaspase-3蛋白(35 kDa)经剪切为17 kDa和12 kDa的活性亚基之后, 可装配成为有活性的caspase-3蛋白, 导致细胞凋亡的发生。实验结果表明gx-50可以一定程度减少氧化损伤引发的PC12细胞凋亡, 并且这一作用受到了JAK2蛋白的特异性抑制剂AG 490的负调控。证实了JAK-STAT通路的活化水平影响了在H2O2诱导的氧化应激环境下, gx-50对于神经元的抗凋亡保护作用。
本实验室前期研究已证明gx-50是一种潜在的α7乙酰胆碱受体激动剂。尼古丁处理过的PC12细胞中, 也有发现招募Jak2蛋白向α7乙酰胆碱受体亚基结合的行为, 而这被认为是其针对神经细胞凋亡的保护机制的一部分[17]。通过激活α7乙酰胆碱受体, 从而进一步激活其下游的JAK-STAT通路, 可能是gx-50在氧化应激环境下发挥对神经元细胞的保护作用的一部分。其具体机制有待于进一步的研究证实。
STAT3对于保持线粒体功能正常发挥起到了重要作用, 并且这一作用是独立于转录机制的[18, 19]
综上所述, 本实验证实了gx-50在Aβ诱导的氧化应激条件下, 对神经元样PC12细胞起到了一定的保护作用。并且gx-50的抗氧化保护作用部分是通过JAK-STAT通路来实现的。对于其更深层的药理学机制, 如gx-50对该通路中其他下游蛋白和基因表达等方面的影响, 以及gx-50激活该通路后引发了哪些级联反应等, 还需要后续研究进一步探讨。
The authors have declared that no competing interests exist.
| [1] |
Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy [J].DOI:10.1196/annals.1427.010 URL PMID: 19076432 [本文引用: 2] 摘要
Abstract Alzheimer's disease (AD) is the most common form of neurodegenerative disease associated with dementia in the elderly. Although the initiating events are still unknown, it is clear that AD, at least in its sporadic form, results from the combination of genetic risk factors with different epigenetic events. Among them, a growing body of evidence suggests that an imbalance between free radical formation and destruction is involved in AD pathogenesis. This concept originally derived from the free radical hypothesis of aging, which states that the age-related accumulation of free radicals results in damaged cell components. The fact that age is a key risk factor in AD provides support for this hypothesis. There is a long list of surrogate markers, which includes lipid, DNA, and protein oxidation, of oxidant stress-mediated injury that have been reported as elevated in the AD brain. Moreover, epidemiologic studies show that dietary intake of natural or synthetic products with a putative antioxidant effect, such as (but not only) vitamin E, reduces the risk of AD. On the other hand, antioxidative intervention studies in animal models of AD-like amyloidosis show a significant reduction in amyloid beta deposition and behavioral improvements. However, a randomized clinical trial of vitamin E supplementation in AD patients shows only a marginal positive effect. Another study reports no effect of vitamin E on the rate of progression of AD in subjects with mild cognitive impairment. This article will review both promises and caveats of the available data and propose future directions to be taken for addressing them.
|
| [2] |
In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy [J].URL PMID: 328613207063181838931172922232222126576808 [本文引用: 1] |
| [3] |
GSK-3/CREB pathway involved in the gx-50's effect on Alzheimer's disease [J].DOI:10.1016/j.neuropharm.2014.02.008 URL PMID: 24565641 [本文引用: 2] 摘要
Aggregation of amyloid-beta (Aβ) fragments is one of the major pathological hallmarks of Alzheimer's disease (AD). Our previous study has demonstrated that a novel compound named N-[2-(3, 4-dimethoxyphenyl) ethyl]-3-phenyl-acrylamide (gx-50) can decrease the accumulation of Aβ oligomers in the cerebral cortex and improve the cognitive abilities in transgenic demented mice. To further study the mechanism of the neuroprotective effect of gx-50 against AD, we employed microarray to investigate the gene expression profile of the primary cultured neurons treated with gx-50 or/and Aβ. Microarray disclosed 351 genes associated with AD in the gx-50 plus Aβ treated group, out of the 22,523 probes. 217 of the 351 genes were significantly up-regulated, 134 of them were down-regulated. The 351 genes were mainly involved in neurotransmission, signal transduction, nervous system development, protein phosphorylation, transcription and apoptosis. By the Onto-pathway analysis, a network involved two molecules – GSK-3, CREB and another two closely linked proteins – AKT, BDNF was discovered. The GSK/CREB pathway was further studied at the gene and protein level both invivo and invitro . Western blot and immunohistochemistry analysis showed that the gx-50 elevated the AKT phosphorylation and inhibited its downstream protein – GSK-3’s activity, then restored the CREB's transcriptional activity, and finally enhanced the expression of the CREB target gene – BDNF. In addition, the real-time PCR results displayed the same tendency. In conclusion, studies in this research indicated that the gx-50 may improve the cognitive ability of AD via the GSK-3/CREB pathway.
|
| [4] |
Survival response of hippocampal neurons under low oxygen conditions induced by hippophae rhamnoides is associated with JAK/STAT signaling [J].DOI:10.1371/journal.pone.0087694 URL PMID: 24516559 [本文引用: 1] 摘要
Janus activated kinase/signal transducers and activators of transcription (JAK/STATs) pathway are associated with various neuronal functions including cell survival and inflammation. In the present study, it is hypothesized that protective action of aqueous extract of Hippophae rhamnoides in hippocampal neurons against hypoxia is mediated via JAK/STATs. Neuronal cells exposed to hypoxia (0.5% O 2 ) display higher reactive oxygen species with compromised antioxidant status compared to unexposed control cells. Further, these cells had elevated levels of pro-inflammatory cytokines; tumor necrosis factor 伪 and interleukin 6 and nuclear factor 魏appa B. Moreover, the expression of JAK1 was found to be highly expressed with phosphorylation of STAT3 and STAT5. Cells treated with JAK1, STAT3 and STAT5 specific inhibitors resulted in more cell death compared to hypoxic cells. Treatment of cells with extract prevented oxidative stress and inflammatory response associated with hypoxia. The extract treated cells had more cell survival than hypoxic cells with induction of JAK1 and STAT5b. Cells treated with extract having suppressed JAK1 or STAT3 or STAT5 expression showed reduced cell viability than the cell treated with extract alone. Overall, the findings from these studies indicate that the aqueous extract of Hippophae rhamnoides treatment inhibited hypoxia induced oxidative stress by altering cellular JAK1, STAT3 and STAT5 levels thereby enhancing cellular survival response to hypoxia and provide a basis for possible use of aqueous extract of Hippophae rhamnoides in facilitating tolerance to hypoxia.
|
| [5] |
Genistein ameliorates beta-amyloid peptide(25-35)-induced hippocampal neuronal apoptosis [J].DOI:10.1016/j.freeradbiomed.2003.10.018 Magsci [本文引用: 1] 摘要
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">β-Amyloid protein (Aβ), a major component of senile plaques of Alzheimer's disease (AD) brain, causes elevation of the intracellular free Ca<sup>2+</sup> level and the production of robust free radicals, both of which contribute greatly to the AD-associated cascade including severe neuronal loss in the hippocampus. Genistein, the most active molecule of soy isoflavones, protects diverse kinds of cells from damage caused by a variety of toxic stimuli. In the present study, we investigated the neuroprotective effect of genistein against Aβ<sub>25–35</sub>-induced apoptosis in cultured hippocampal neurons, as well as the underlying mechanism. Aβ<sub>25–35</sub>-induced apoptosis, characterized by decreased cell viability, neuronal DNA condensation, and fragmentation, is associated with an increase in intracellular free Ca<sup>2+</sup> level, the accumulation of reactive oxygen species (ROS), and the activation of caspase-3. All these phenotypes induced by Aβ<sub>25–35</sub> are reversed by genistein. Our results further show that at the nanomolar (100 nM) level, genistein protects neurons from Aβ<sub>25–35</sub>-induced damage largely via the estrogen receptor-mediated pathway, and at the micromolar (40 μM) level, the neuroprotective effect of genistein is mediated mainly by its antioxidative properties. Our data suggest that genistein attenuates neuronal apoptosis induced by Aβ<sub>25–35</sub> via various mechanisms.</p>
|
| [6] |
Oxidants, oxidative stress and the biology of ageing [J].DOI:10.1038/35041687 URL PMID: 11089981 [本文引用: 1] 摘要
Discusses the relationship of oxidants and oxidative stress to the biology of aging. Explanation of how reactive oxygen species (ROS) are generated; How the cell responds to oxidative stress, and how these responses change with age; Discussion of the challenges associated with the potential development of anti-aging therapies.
|
| [7] |
Oxidative stress and ageing in Caenorhabditis elegans [J].
Mutations in the age-1 gene double both the mean and maximum life span of Caenorhabditis elegans. They also result in an age-specific increase of catalase and Cu/Zn superoxide dismutase activity levels. The higher superoxide dismutase activity levels in age-1 mutants confer hyperresistance to the superoxide-anion-generating drug paraquat. The rate of superoxide anion production by microsome fractions declines linearly with age in age-1(+) worms, but, after an initial decline, is stabilized at a higher level in senescent age-1 mutant nematodes. These results clearly show that oxidative stress resistance and potential life span are correlated in this organism, and they suggest that the natural product of age-1 either directly or indirectly downregulates the activities of several other genes as a function of age.
|
| [8] |
Aging and resistance to oxidative damage in Caenorhabditis elegans [J].DOI:10.1073/pnas.90.19.8905 URL PMID: 8415630 [本文引用: 1] 摘要
The dauer larva state and the age-1 mutation, both of which extend life-span in Caenorhabditis elegans, were tested for hyperresistance to cellular damage that may be relevant to aging. The age-1 strain TJ401 displayed hyperresistance to oxidative stress relative to its parental strain. The activities of two enzymes that protect cells from oxidative damage, superoxide dismutase (SOD) and catalase, showed an age-dependent increase in mutant animals, which was not seen in the parental strain. These increases in activities paralleled the time course of the hyperresistance. The results are consistent with the age-1 gene product functioning as a negative regulator of SOD and catalase activities. In wild-type and age-1 dauer larvae, elevated levels of SOD activity, but not of catalase activity, were present when compared with young adults. The common increase in SOD activity prompted cloning the C. elegans Cu/Zn SOD gene. Its position on the physical map of the genome was in the region to which the age-1 gene has been genetically mapped, but it is unlikely that a mutation at the SOD locus confers the Age phenotype. Results support the free radical theory of aging by suggesting that the increased resistance to oxidative stress may be among the causes of increased longevity in both strain TJ401 and in the dauer larva.
|
| [9] |
Genetic analysis of ageing:role of oxidative damage and environmental stresses [J].DOI:10.1038/ng0596-25 URL PMID: 8673100 [本文引用: 1] 摘要
Evolutionary theory predicts substantial interspecific and intraspecific differences in the proximal mechanisms of . Our goal here is to seek evidence for common ('public') mechanisms among diverse organisms amenable to genetic analysis. Oxidative damage is a candidate for such a public mechanism of . Long-lived strains are relatively resistant to different environmental stresses. The extent to which these stresses produce oxidative damage remains to be established.
|
| [10] |
Oxidative damage during aging targets mitochondrial aconitase [J].DOI:10.1073/pnas.94.21.11168 URL PMID: 9326580 [本文引用: 1] 摘要
Oxidative damage during aging targets mitochondrial aconitase YAN LJ. Proc. Natl Acad. Sci. 94, 11168-11172, 1997
|
| [11] |
Amyloid precursor protein-mediated free radicals and oxidative damage:Implications for the development and progression of Alzheimer's disease [J].
|
| [12] |
A novel drug candidate for Alzheimer's disease treatment:gx-50 derived from Zanthoxylum Bungeanum [J].DOI:10.3233/JAD-121831 URL PMID: 23186988 [本文引用: 3] 摘要
This study focused on a promising drug candidate, N-[2-(3,4-dimethoxyphenyl)ethyl]-3-phenyl-acrylamide (gx-50), a compound extracted from Sichuan pepper (Zanthoxylum Bungeanum), to determine whether it would be an effective therapeutic for Alzheimer's disease (AD) via biological experiments. In vivo, we determined the pharmacokinetic profile of gx-50 and evaluated the effect of gx-50 on the cognitive abilities of amyloid-β protein precursor transgenic (AβPP-Tg) mice by Morris water maze testing. In addition, we examined the effects of gx-50 on amyloid-β (Aβ) oligomers in the brains of AβPP-Tg mice by immunohistochemistry. In vitro, we observed a direct effect of gx-50 on Aβ oligomers by atomic force microscopy, detected the neuroprotective effects of gx-50 by western blotting and cell apoptosis assays, and measured its effects on intracellular calcium currents by laser confocal microscopy. Experiments in vivo showed that gx-50 could penetrate the blood brain barrier and improve the cognitive abilities of mice. Moreover, gx-50 treatment decreased the accumulation of Aβ oligomers in the cerebral cortex. The results in vitro demonstrated that gx-50 could disassemble Aβ oligomers, inhibit Aβ-induced neuronal apoptosis and apoptotic gene expression, and reduce neuronal calcium toxicity. These results strongly suggest that gx-50 is a potential candidate drug for treating AD.
|
| [13] |
The suppressive effects of gx-50 on Aβ-induced chemotactic migration of microglia [J]. |
| [14] |
Targeting the JAK2/STAT3 axis in Alzheimer's disease [J].DOI:10.1517/14728220903213426 URL PMID: 19663649 [本文引用: 1] 摘要
Background: Amyloid 尾 (A尾) has long been implicated in the pathogenesis of Alzheimer's disease (AD). Little is known, however, about the intracellular events in neurons which lead to memory loss related to AD. Focusing on the fact that an AD-specific neuroprotective peptide named humanin (HN) inhibits AD-related neurotoxicity by activating the JAK2/STAT3 signaling axis, we recently found that age- and disease-dependent deterioration in the JAK2/STAT3 axis plays a critical role in the pathogenesis of AD. Objective/methods: Here we summarize the neuroprotective effect of HN and its derivative, named colivelin (CLN), and also review the roles of the JAK2/STAT3 axis in memory impairment related to AD. Results/conclusions: The JAK2/STAT3 axis is a major transducer of HN-mediated neuroprotective activity. A尾-dependent inactivation of the JAK2/STAT3 axis in hippocampal neurons causes cholinergic dysfunction via pre- and post-synaptic mechanisms, which leads to memory impairment related to AD. This provides not o...
|
| [15] |
Amyloid-β causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons [J].DOI:10.1038/mp.2008.105 URL [本文引用: 1] 摘要
Elevation of intracranial soluble amyloid-0205 (A0205) levels has been implicated in the pathogenesis of Alzheimer's disease (AD). Intracellular events in neurons, which lead to memory loss in AD, however, remain elusive. Humanin (HN) is a short neuroprotective peptide abolishing A0205 neurotoxicity. Recently, we found that HN derivatives activate the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling axis. We here report that an HN derivative named colivelin completely restored cognitive function in an AD model (Tg2576) by activating the JAK2/STAT3 axis. In accordance, immunofluorescence staining using a specific antibody against phospho- (p-) STAT3 revealed that p-STAT3 levels in hippocampal neurons age-dependently decreased in both AD model mice and AD patients. Intracerebroventricular administration of A0205109恪42 downregulated p-STAT3 whereas passive immunization with anti-A0205 antibody conversely restored hippocampal p-STAT3 levels in Tg2576 mice, paralleling the decrease in the brain A0205 burden. A0205109恪42 consistently modulated p-STAT3 levels in primary neurons. Pharmacological inhibition of the JAK2/STAT3 axis not only induced significant loss of spatial working memory by downregulating an acetylcholine-producing enzyme choline acetyltransferase but also desensitized the M1-type muscarinic acetylcholine receptor. Thus, we propose a novel theory accounting for memory impairment related to AD: A0205-dependent inactivation of the JAK2/STAT3 axis causes memory loss through cholinergic dysfunction. Our findings provide not only a novel pathological hallmark in AD but also a novel target in AD therapy.Molecular Psychiatry (2009) 14, 20609恪222; doi:10.1038/mp.2008.105; published online 23 September 2008
|
| [16] |
Loss of STAT3 in mouse embryonic fibroblasts reveals its Janus-like actions on mitochondrial function and cell viability [J].DOI:10.1016/j.cyto.2013.12.006 URL PMID: 24548419 [本文引用: 1] 摘要
STAT3 has been implicated in mitochondrial function; however, the physiological relevance of this action is not established. Here we studied the importance of STAT3 to the cellular response to stimuli, TNFα and serum deprivation, which increase mitochondrial reactive oxygen species (ROS) formation. Experiments were performed using wild type (WT) and STAT3 knockout (KO) mouse embryonic fibroblasts (MEF). Both WT and STAT3 KO MEF expressed similar levels of tumor necrosis factor receptor 1 (TNFR1) and exhibited comparable IκBα degradation with TNFα. However, in the absence of STAT3 nuclear accumulation of NFκB p65 with TNFα was attenuated and induction of the survival protein c-FLIPL was eliminated. Nonetheless, WT MEF were more sensitive to TNFα-induced death which was attributed to necrosis. Deletion of STAT3 decreased ROS formation induced by TNFα and serum deprivation. STAT3 deletion was associated with lower levels of complex I and rates of respiration. Relative to WT cells, mitochondria of STAT3 KO cells released significantly more cytochrome c in response to oxidative stress and had greater caspase 3 cleavage due to serum deprivation. Our findings are consistent with STAT3 being important for mitochondrial function and cell viability by ensuring mitochondrial integrity and the expression of pro-survival genes.
|
| [17] |
Janus kinase 2, an early target of α7 nicotinic acetylcholine receptor-mediated neuroprotection against Aβ-(1-42)amyloid [J].DOI:10.1074/jbc.M204610200 URL [本文引用: 1] 摘要
The molecular mechanisms of α7 nicotinic acetylcholine receptor (nAChR)-mediated neuroprotection remain unclear. In this study we provide evidence that nicotine stimulation of α7 nAChR transduces signals to phosphatidylinositol 3-kinase and Akt via Janus kinase 2 (JAK2) in a cascade, which results in neuroprotection. Exposure to β-amyloid results in the activation of the apoptotic enzyme caspase-3 and cleavage of the DNA-repairing enzyme poly-(ADP-ribose) polymerase. This cascade is inhibited by nicotine through JAK2 activation, and these effects are blocked by preincubation with the JAK2-specific inhibitor AG-490. We also found that pretreatment of cells with angiotensin II blocks the nicotine-induced activation of JAK2 via the AT 2 receptor and completely prevents α7 nAChR-mediated neuroprotective effects further suggesting a pivotal role for JAK2. These findings identify novel mechanisms of receptor interactions relevant to neuronal viability and suggest novel therapeutic strategies to optimize neuroprotection.
|
| [18] |
Multi-tasking:nuclear transcription factors with novel roles in the mitochondria [J].DOI:10.1016/j.tcb.2012.05.001 URL PMID: 22705015 [本文引用: 1] 摘要
Abstract Coordinated responses between the nucleus and mitochondria are essential for the maintenance of homeostasis. For over 15 years, pools of nuclear transcription factors (TFs), such as p53 and nuclear hormone receptors, have been observed in the mitochondria. The contribution of the mitochondrial pool of these TFs to their well-defined biological actions is in some cases clear and in others not well understood. Recently, a small mitochondrial pool of the TF signal transducer and activator of transcription factor 3 (STAT3) was shown to modulate the activity of the electron transport chain (ETC). The mitochondrial function of STAT3 encompasses both its biological actions in the heart as well as its oncogenic effects. This review highlights advances in our understanding of how mitochondrial pools of nuclear TFs may influence the function of this organelle. Copyright 漏 2012 Elsevier Ltd. All rights reserved.
|
| [19] |
Cytoprotection by the modulation of mitochondrial electron transport chain:the emerging role of mitochondrial STAT3 [J]. |
| [20] |
Mitochondrial metabolism of reactive oxygen species [J].DOI:10.1007/s10541-005-0102-7 URL PMID: 15807660 摘要
Oxidative stress is considered a major contributor to etiology of both "normal" senescence and severe pathologies with serious public health implications. Mitochondria generate reactive oxygen species (ROS) that are thought to augment intracellular oxidative stress. Mitochondria possess at least nine known sites that are capable of generating superoxide anion, a progenitor ROS. Mitochondria also possess numerous ROS defense systems that are much less studied. Studies of the last three decades shed light on many important mechanistic details of mitochondrial ROS production, but the bigger picture remains obscure. This review summarizes the current knowledge about major components involved in mitochondrial ROS metabolism and factors that regulate ROS generation and removal. An integrative, systemic approach is applied to analysis of mitochondrial ROS metabolism, which is now dissected into mitochondrial ROS production, mitochondrial ROS removal, and mitochondrial ROS emission. It is suggested that mitochondria augment intracellular oxidative stress due primarily to failure of their ROS removal systems, whereas the role of mitochondrial ROS emission is yet to be determined and a net increase in mitochondrial ROS production in situ remains to be demonstrated.
|

| 沪交ICP备05221 版权所有:《上海交通大学学报(农业科学版)》编辑部 主管单位:中华人民共和国教育部 主办单位:上海交通大学 出版单位:上海交通大学学报编辑部 地址:上海市七莘路2678号 上海交通大学七宝校区36号信箱 邮政编码:201101 电话:021-64789728 电子邮件:xuebao@sjtu.edu.cn |
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}