|
||
|

|
杨昊 , 张瑞, 黄丹枫
, 张瑞, 黄丹枫
YANG Hao, Pablo Gonzalez Perez
文献标识码: 1671-9964(2016)01-0036-09
文章编号: 1671-9964(2016)01-0036-09
收稿日期: 2014-12-22
网络出版日期: 2016-01-20
版权声明: 2016 上海交通大学期刊中心 版权所有
基金资助:
作者简介:
作者简介: 杨昊(1988-), 男, 硕士生, 研究方向:菜田土壤微生物多样性, E-mail: yanghaoiloveu@163.com;
通讯作者: 黄丹枫(1956-), 女, 教授, 博士生导师, 研究方向:园艺作物生理生态, E-mail:hdf@sjtu.edu.cn; 蔡保松(1971-), 男, 副教授, 硕士生导师, 研究方向:园艺作物生理生态, E-mail:czstl@sjtu.edu.cn
展开
摘要
通过模拟试验, 利用聚合酶链式反应-变性梯度凝胶电泳法(PCR-DGGE), 研究了施入不同碳氮有机物料(秸秆、苜蓿、有机肥、尿素)56 d后, 有机生产系统菜田土壤细菌群落结构的特征。结果表明, 常规和有机生产系统土壤的细菌群落结构有明显差异。由DGGE图谱Shannon-Wiener多样性指数分析得知, 有机背景处理的细菌多样性整体高于常规背景处理, 且有机生产系统土壤加秸秆处理(OS)多样性最高, 加入尿素后细菌多样性降低, 相反, 加入苜蓿后细菌多样性升高。非加权组平均法(UPGMA)聚类分析将常规和有机背景土壤分为两大族群。DGGE条带测序和系统进化树表明, 30个条带归属为Proteobacteria、Acidobacteria、Actinobacteria、Firmicutes、Verrucomicrobia。常规土壤加苜蓿(CA)处理出现的特征性条带B13与有机背景处理的共有条带B28分别与Bacillus属和Pseudomonas假单胞菌属同源性最高。
关键词:
Abstract
Through the simulation test, PCR-DGGE method was used to investigate the effects of different carbon and nitrogen organic materials on bacterial community structure of organic vegetable production system soil.Results showed that bacterial community structure of conventional and organic production system soils were significantly different.By analyzing the Shannon-Wiener diversity index, overall the bacterial diversity of organic background soil treatments was higher than conventional background soil treatments.The bacterial diversity of organic production system soil with straw(OS)was the highest.Bacterial diversity decrease or increase after adding urea or alfalfa respectively.Conventional and organic production system soils were divided into two ethnic clusters by clustering analysis.Phylogenetic tree showed that the 30 closely related species were attributed to Proteobacteria, Acidobacteria, Actinobacteria, Firmicutes, Verrucomicrobia.The band B13 of CA(conventional production system soil with alfalfa)and the common band B28 of organic background treatment had the highest homology with Bacillus and Pseudomonas respectively.
Keywords:
土壤中大多数微生物无法在实验室培养, 随着技术的发展, 越来越多的学者开始从分子水平进行微生物多样性的研究, 并因此逐渐形成了土壤微生物分子生态学的研究方向, 这丰富了人们对土壤生态系统中微生物的认识。土壤微生物是土壤生态系统中的重要组成部分, 参与土壤的大多数生化过程, 在农业生产过程中, 施入肥料可影响土壤微生物的群落结构。有研究结果指出, 长期施用有机肥后土壤细菌多样性要高于化肥处理[1]; 也有研究指出, 长期施用化肥增加了细菌的多样性, 而长期施用有机肥对细菌多样性的影响较小[2]; 也有研究报道长期施肥并没有显著改变土壤中的细菌群落[3]。并有研究认为, 施用有机肥增加的细菌主要是由于有机肥促进了土壤里土著细菌的增长[4]。同时秸秆还田可以增加土壤中细菌的数量[5]。一般认为, 肥料是通过直接或间接影响土壤的理化性质, 改变土壤的微生境, 进而影响土壤微生物的群落结构。但是关于施肥对土壤微生物影响的研究多在大田环境下进行, 然而土壤微生物容易受气候、温度、大气等环境因素的影响, 室内模拟试验可排除这些因素的影响, 相关研究鲜有报道。本试验即在室内模拟条件下, 采用PCR-DGGE(聚合酶链式反应-变性梯度凝胶电泳法)的分子技术研究不同外源有机物料对有机菜田土壤细菌多样性的影响, 以期为有机农业生产合理施肥提供理论依据。
试验土壤取自上海崇本堂农场, 位于上海市松江区叶榭镇(30°57'N, 121°21'E), 北距黄浦江约2.5 km, 南距杭州湾约23 km。属亚热带湿润气候, 海拔4 m, 年平均降雨量1 550 mm, 年平均气温为17.5 ℃, 年平均日照为1 778 h。该农场有机生产系统露地种植始于2002年, 已有12年种植历史, 正常情况下每年种植3茬, 年有机肥施用量为112.5 t/hm2; 常规生产系统露地种植每年3茬, 年化肥使用量为复合肥(N∶P∶K=14∶16∶15)4 000 kg/hm2, 尿素1 200 kg/hm2。2013年3月(当季休耕), 分别在有机和常规生产系统露地种植区, 选取15 m×50 m的地块, 按照S型8点取样方法, 用管型土钻采集这8个点的0~20 cm土壤, 然后分别进行混合, 有机和常规生产系统土壤各采集40 kg, 最后在实验室对这些土壤样品进行后续处理。
分别将上述采集的两类土壤过2 mm孔径的筛子, 除去可见植物残渣, 然后分别混合均匀, 测量混匀后土壤的含水量和最大持水量。两类土壤的理化性质见表1。试验处理共有8个, 分别为常规土壤(C)、常规土壤+尿素(CU)、常规土壤+苜蓿(CA)、有机土壤(O)、有机土壤+有机肥(OO)、有机土壤+尿素(OU)、有机土壤+秸秆(OS)、有机土壤+苜蓿(OA), 其中尿素(45.5% N, 20.0% C, C/N=0.44)、苜蓿(3.8% N, 43.3% C, C/N=11.39)、有机肥(2.0% N, 31.6% C, C/N=15.8)、秸秆(1.4% N, 40.5% C, C/N=28.93)按照0.1 mg/g干土进行添加, 添加前把苜蓿和秸秆研磨至5~10 mm长度左右, 以增大其与土壤接触的表面积, 添加完后混合均匀。然后将大约250 g处理后的土壤(转换为干土的重量)放入580 mL的梅森瓶, 每个处理3次重复, 梅森瓶盖子打孔, 以保障通气, 对每个重复(包括瓶子)进行称重并记下重量。然后将其放入人工气候室, 土壤含水量保持在最大持水量的50%, 以保证在土壤湿润的同时有足够的氧气, 土壤培养的过程中根据之前称重的重量变化决定加水量, 人工气候室温度设置为(20±1) ℃, 全黑暗条件培养。培养56 d后把每个重复的土壤全部取出, 然后分为三部分:一部分自然风干后保存, 一部分保存于-20 ℃, 一部分保存于-80 ℃进行后续试验。
1.3.1 土壤微生物总DNA提取和PCR扩增
(1) 土壤微生物总DNA提取。用于总DNA提取的土壤样品由各处理3个重复土样均匀混合而成。采用E.Z.N.A.TM Soil DNA Kit D5625-01(OMEGA, 美国)试剂盒, 按照试剂盒操作说明对土壤微生物DNA进行提取和纯化, 提取的土壤DNA浓度及纯度使用微量紫外分光光度计NanoDrop1000(Thermo Fisher Scientific, 美国)进行鉴定。然后将DNA稀释20倍用于后续PCR扩增实验。
(2) 土壤细菌的PCR扩增。采用细菌16S rDNA基因具有特异性的PCR引物1401r[6](5'-CGGTGTGTACAAGACCC-3')和GC-968(5'-GCclamp-AACGCGAAGAACCTTAC-3')(GC-clamp序列为:CCGCCGCGCGGCGGGCGGGG-CGGGGGCACGGGG)对土壤DNA进行扩增。PCR反应体系为50 μL, 其中包括:1 μL模板DNA、5 μL的10×PCR Buffer、4 μL的dNTP Mixture(各2.5 mmol/L)、10 μmmol/L正反向引物各1 μL、0.5 μL的5 U/μL TaqDNA聚合酶(Takara, 大连)、37.5 μL灭菌双蒸水。PCR反应条件为:94 ℃预变性5 min; 35个循环为94 ℃ 30 s, 57 ℃ 45 s, 72 ℃ 45 s; 最后在72 ℃下延伸10 min。PCR反应产物用1%的琼脂糖凝胶电泳检测, 4 ℃保存PCR反应产物, PCR扩增片段长约为440 bp。
1.3.2 PCR产物的变性梯度凝胶电泳(DGGE)分析
使用Bio-Rad公司的DCodeTM Universal Mutation Detection System对细菌PCR扩增产物进行DGGE电泳分析。聚丙烯酰胺凝胶浓度为8%, 变性梯度为35%~60%(100%变性剂成分为:37.5∶1的40% Acrylamide/ Bis 20 mL、50×TAE buffer 2 mL、去离子甲酰胺40 mL、尿素42 g、灭菌纯水补齐至100 mL)。将30 μL的PCR产物和6 μL的6×Loading buffer充分混匀后加入上样孔, 然后将胶板致于1×TAE电泳液中, 在65 V电压和60 ℃恒温条件下电泳15 h。电泳完毕后, 取出胶板在避光条件下进行银染[7]。最后用UMAX Powerlook 2100XL-USB扫描仪获取图像。
1.3.3 DGGE条带的切割与测序
从DGGE凝胶上小心切取分离的条带, 将其放入1.5 mL的离心管中, 用100 μL的无菌双蒸水将条带冲洗3遍后, 加入20 μL的灭菌双蒸水, 用灭菌的枪头将其捣碎, 沸水浴5 min使凝胶融化, 4 ℃过夜保存。次日取1 μL作为模版, 以1401r和不带GC-clamp的968为引物按照1.3.1(2)的反应程序再次进行PCR扩增。用1%琼脂糖对PCR产物电泳后进行切胶, 使用SanPrep柱式DNA胶回收试剂盒(上海生工生物工程股份有限公司)对切割后的条带进行回收纯化。纯化产物与pMD19-T载体(Takara, 大连)进行连接, 然后转化到E.coli DH5α感受态细胞中, 在含有氨苄青霉素、X-gal(5-溴-4-氯-3-吲哚-β-D-半乳糖苷)、IPTG(异丙基硫代半乳糖苷)的LB固体培养基上选择具有氨苄青霉素抗性的白色转化子。白色转化子在含有氨苄青霉素的LB液体培养基中经过培养后, 用T载体通用引物M13F和M13R进行阳性检测后进行测序, 测序工作由上海桑尼生物科技有限公司完成。
1.3.4 微生物群落基因序列分析
利用NCBI的BLAST程序对所得序列进行同源性比较, 下载与本试验所得序列相似性最高的序列, 并将所得序列提交到GenBank数据库中。使用MEGA软件对本试验所得序列和相似性最高的序列进行比对, 采用邻接法选用p-distance模型进行系统发育树的构建。
利用Quantity One分析软件对DGGE图谱进行数字化处理分析。土壤微生物多样性采用Shannon-Wiener指数(H)、Margalef丰富度指数(D)、Pielou均匀度指数(E)进行评价, 以上指数计算公式同文献[3], 数据用Microsoft Excel 2007进行处理, 统计与显著性检测利用SPSS 19.0软件进行。
细菌DGGE指纹图谱结果如图1所示。
从图中我们可以看出无论是常规生产系统还是有机生产系统的土壤, 都出现了共有条带B7、B11、B14、B15、B21、B26、B39、B43, 说明了常规生产和有机生产系统土壤有相同的细菌存在, 然而共有条带B7、B26却在条带的亮度上有显著差别, 说明常规和有机背景土壤尽管存在相同的细菌, 但是在细菌的相对生物量上也有差别。在常规生产系统土壤背景下的处理C、CU、CA中出现了B2、B8、B9、B10等共有条带, 而在有机生产系统背景下的处理O、OO、OU、OS、OA中则出现了B30、B32、B35等共有条带, 说明在常规和有机生产系统背景下的土壤存在不同的细菌群落。在常规生产背景土壤中, 加入苜蓿的处理CA出现了特征性条带B13, 并且B1条带在处理CA中明显出现, 条带B8在处理C中最亮, 在处理CU和CA中亮度变弱, 说明常规生产背景的土壤, 在加入不同的肥料(尿素和苜蓿)后, 对土壤的细菌产生了不同的影响。在有机生产背景土壤中, 加入秸秆的处理OS出现了特征性条带B31、B18, 加入苜蓿的处理OA出现了特征性条带B29, 同时在有机生产背景土壤处理O、OO、OU、OS出现的共有条带B1和B24, 处理OA却没有出现, 这也说明, 有机生产背景的土壤, 加入不同肥料后, 对土壤的细菌也可产生不同的影响。
表1 供试土壤基本理化性质
Tab.1 The properties of the tested soils
| 土壤类型 Soil type | pH | 全碳/ (g kg-1) Total C | 全氮/ (g·kg-1) Total N | C / N | 可溶性有机氮 / (mg·kg-1) SON | 硝态氮/ (mg·kg-1) Nitrate | 铵态氮/ (mg·kg-1) Ammonium | 微生物量碳/ (mg·kg-1) MBC | 微生物量氮/ (mg·kg-1) MBN |
|---|---|---|---|---|---|---|---|---|---|
| 有机土壤 Organic soil | 6.80±0.13 | 26.2±3.4 | 3.1±0.03 | 8.4±1.1 | 94.6±9.6 | 42.06±7.78 | 3.35±0.37 | 75.6±13.1 | 18.5±3.8 |
| 常规土壤 Conventional soil | 7.10±0.09 | 19.2±2.5 | 2.2±0.05 | 8.7±1.0 | 59.1±6.4 | 68.88±13.60 | 3.71±0.39 | 65.2±13.7 | 7.9±1.4 |
图1 土壤细菌群落的PCR-DGGE图谱C为常规土壤; CU为常规土壤+尿素; CA为常规土壤+苜蓿; O为有机土壤; OO为有机土壤+有机肥; OU为有机土壤+尿素; OS为有机土壤+秸秆; OA为有机土壤+苜蓿
Fig.1 PCR-DGGE profile of soil bacterial community structureC: Conventional soil; CU:Conventional soil + urea; CA:Conventional soil + alfalfa; O:Organic soil; OO:Organic soil + organic fertilizer; OU:Organic soil + urea; OS: Organic soil +straw; OA:Organic soil + alfalfa
对供试土壤中细菌多样性指数(H), 丰富度指数(S)和均匀度指数(E)进行综合分析, 结果如表2所示。从表中可看出, 多样性指数和丰富度指数最高的处理是OS处理, 其值分别为3.002和3.568; 最低的处理为CU, 其值分别为2.600和2.409。在常规种植背景下不同处理多样性指数的高低为CA>C>CU, 加入苜蓿后细菌多样性指数明显提高, 加入尿素后细菌多样性指数和对照几乎保持一样。有机生产背景下不同处理多样性指数表现出OS>OA>O>OU>OO的规律, 加入苜蓿和秸秆后, 土壤细菌多样性指数比对照O增加较多, 加入尿素和有机堆肥后, 土壤细菌多样性指数有所下降, 但不明显。并且结果表明, 有机生产背景处理的多样性指数和丰富度指数整体高于常规生产背景的处理。
表2 不同处理土壤细菌群落多样性、丰富度和均匀度指数
Tab.2 Shannon-Wiener(H), Margalef(D)and Evenness(E)indices of soil bacterial communities in the different treatments
| 处理 Treatment | 多样性指数(H) Diversity index | 丰富度指数(D) Richness | 均匀度指数(E) Evenness |
|---|---|---|---|
| C | 2.611 | 2.523 | 0.942 |
| CU | 2.600 | 2.409 | 0.938 |
| CA | 2.777 | 2.920 | 0.943 |
| O | 2.855 | 3.141 | 0.938 |
| OO | 2.843 | 3.150 | 0.934 |
| OU | 2.848 | 3.100 | 0.936 |
| OS | 3.002 | 3.568 | 0.944 |
| OA | 2.870 | 3.305 | 0.943 |
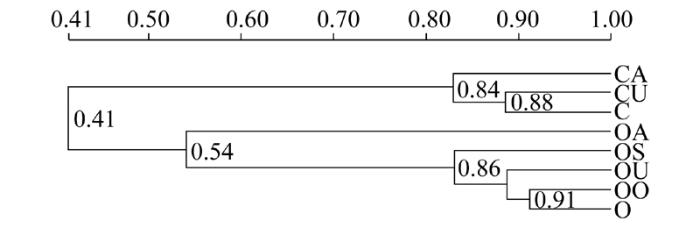
细菌聚类结果如图2所示。8个处理聚类为二大族群, C、CU、CA为第一族群, OA、OS、OU、OO、O为第二族群, 即常规种植背景和有机种植背景土壤聚类为二大族群, 说明常规和有机土壤细菌群落存在明显差异。在第一族群里面C和CU聚类为一个亚族群, 添加苜蓿的处理CA单独列为一支, 说明加入苜蓿可对常规土壤细菌群落结构产生影响。在第二大族群里面O和OO聚类为一个亚族群, 加入不同肥料的处理OU、OS、OA分别单独列为一支, 说明在有机种植背景下, 添加尿素、秸秆、苜蓿可对细菌的群落结构产生不同影响。
将细菌DGGE图谱上出现的共有和特殊条带进行切胶回收测序, 共得到30条DGGE条带, 将序列提交至GenBank数据库, 利用NCBI的BLAST程序进行序列同源性比较, 将同源性最高的序列下载, 结果如表3。从表3可以看出, 所测序列与GenBank数据库中的16S rRNA序列, 同源性B10为92%, B13为94%, B18为93%, B26为94%, B32为91%, B44为95%, B46为94%, 其他序列同源性都在97%以上, 同源性很高。
图2 细菌DGGE图谱UPGMA聚类分析C为常规土壤; CU为常规土壤+尿素; CA为常规土壤+苜蓿; O为有机土壤; OO为有机土壤+有机肥; OU为有机土壤+尿素; OS为有机土壤+秸秆; OA为有机土壤+苜蓿
Fig.2 Cluster analysis of bacterial DGGE with UPGMAC:Conventional soil; CU:Conventional soil + urea; CA:Conventional soil + alfalfa; O:Organic soil; OO:Organic soil + organic fertilizer; OU:Organic soil + urea; OS:Organic soil +straw; OA:Organic soil + alfalfa
表3 细菌DGGE图谱条带的序列比对结果
Tab.3 Sequence alignment of bands in bacterial DGGE profile
| 条带 Band | 登录号 Genebank Accession No. | 序列长度 Sequence lehgth(bp) | 系统分类 Phylogenetic affiliation | 相似菌株 Closest Identified Relative (Genebank Accession No.) | 同源性 Homology(%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| B1 | KM281157 | 435 | Firmicutes; Clostridiaceae; Clostridium. | Uncultured Clostridium sp.clone T3-69 16S ribosomal RNA gene, partial sequence(KM200463) | 99% | ||||
| B2 | KM281158 | 434 | Acidobacteria; Acidobacteriales. | Uncultured Acidobacteriales bacterium clone E2006TS6.44 16S ribosomal RNA gene, partial sequence(GU983317) | 99% | ||||
| B5 | KM281161 | 434 | Acidobacteria; | Uncultured Acidobacteria bacterium clone AKYG1109 16S ribosomal RNA gene, partial sequence(AY921847) | 99% | ||||
| B7 | KM281163 | 438 | Uncultured bacterium(HQ119238) | 99% | |||||
| B8 | KM281164 | 434 | Acidobacteria; | Uncultured Acidobacteria bacterium clone HG-B02190 16S ribosomal RNA gene, partial sequence(JN409248) | 99% | ||||
| B9 | KM281165 | 434 | Uncultured bacterium clone WIF2 16S ribosomal RNA gene, partial Sequence(HQ166676) | 100% | |||||
| B10 | KM281166 | 437 | Uncultured bacterium(JF708475) | 92% | |||||
| B11 | KM281167 | 435 | Uncultured bacterium clone CP63 16S ribosomal RNA gene, partial sequence(JX133269) | 100% | |||||
| B12 | KM281168 | 437 | Uncultured bacterium(JF708475) | 100% | |||||
| B13 | KM281169 | 436 | Firmicutes; Bacillaceae; Bacillus. | Uncultured Bacillus sp.clone SGR274 16S ribosomal RNA gene, partial sequence(JQ793581) | 94% | ||||
| B14 | KM281170 | 434 | Proteobacteria; Rhodocyclales; Rhodocyclaceae. | Uncultured Rhodocyclaceae bacterium partial 16S rRNA gene, clone SBROB5_44(LN555043) | 98% | ||||
| B15 | KM281171 | 436 | Proteobacteria; Alphaproteobacteria; | Uncultured alpha proteobacterium(JN038900) | 99% | ||||
| B16 | KM281172 | 435 | Uncultured bacterium clone MCzcultivatedPlana46 16S ribosomal RNA gene, partial sequence(HM444534) | 99% | |||||
| B18 | KM281174 | 438 | Acidobacteria; | Uncultured Acidobacteria(JN409214) | 93% | ||||
| B19 | KM281175 | 439 | Actinobacteria; Intrasporangiaceae; Humibacillus. | Humibacillus xanthopallidus gene for 16S rRNA, partial sequence, strain:S32487(AB648995) | 99% | ||||
| B21 | KM281177 | 431 | Uncultured bacterium isolate DGGE gel band CB-9 16S ribosomal RNA gene, partial sequence(KJ667548) | 99% | |||||
| B24 | KM281180 | 433 | Actinobacteria; Micromonosporaceae; Planosporangium. | Planosporangium thailandense strain HSS8-18 16S ribosomal RNA gene, partial sequence(NR_108119) | 98% | ||||
| B26 | KM281181 | 436 | Proteobacteria; Xanthomonadales; Sinobacteraceae. | Uncultured Sinobacteraceae bacterium partial 16S rRNA gene, clone SBROB5_36(LN555035) | 94% | ||||
| B28 | KM281183 | 435 | Proteobacteria; Pseudomonadaceae; Pseudomonas. | Pseudomonas sp.CC-MHH0089 16S ribosomal RNA gene, partial sequence(KJ720680) | 99% | ||||
| B29 | KM281184 | 431 | Uncultured bacterium clone BacC-s_023 16S ribosomal RNA gene, partial sequence(EU335200) | 98% | |||||
| B30 | KM281185 | 434 | Uncultured bacterium partial 16S rRNAgene, clone SICT499_N9D2_16S_B(LN570594) | 99% | |||||
| B31 | KM281186 | 435 | Uncultured bacterium(DQ973988) | 97% | |||||
| B32 | KM281187 | 434 | Uncultured bacterium(GU320659) | 91% | |||||
| B34 | KM281188 | 433 | Actinobacteria; Nocardioidaceae; Marmoricola. | Marmoricola sp.M57 16S ribosomal RNA gene, partial sequence(KC464831) | 99% | ||||
| B35 | KM281189 | 435 | Acidobacteria; | Uncultured Acidobacteria(JN408907) | 99% | ||||
| B39 | KM281192 | 433 | Actinobacteria; Propionibacterineae; Nocardioidaceae; Marmoricola. | Uncultured Marmoricola sp.clone BG1-39 16S ribosomal RNA gene, partial sequence(JX079124) | 97% | ||||
| B42 | KM281194 | 434 | Verrucomicrobia; Verrucomicrobiales; Verrucomicrobiaceae. | Uncultured Verrucomicrobiaceae(FJ542838) | 98% | ||||
| B43 | KM281195 | 434 | Uncultured bacterium(JX133426) | 100% | |||||
| B44 | KM281196 | 422 | Uncultured bacterium(DQ499290) | 95% | |||||
| B46 | KM281198 | 437 | Uncultured bacterium isolate DGGE gel band CB-16 16S ribosomal RNA gene, partial sequence(KJ667555) | 94% | |||||
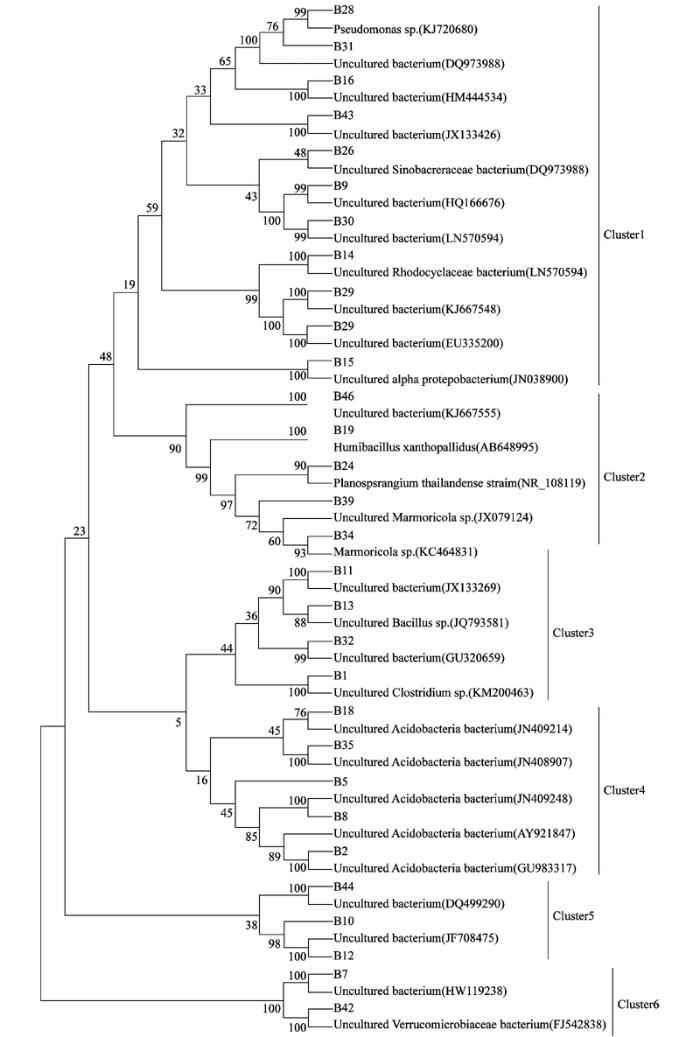
通过MEGA 5软件构建系统发育树, 结果如图3。从系统发育树可见细菌群落的组成多样性, 30个条带与其相似菌株被分为6大簇。其中第1簇归类为Proteobacteria门, 第2簇归类为Actinobacteria门, 第3簇归类为Firmicutes门, 第4族归类为Acidobacteria门, 第5簇为未确定归属的细菌, 第6簇归类为Verrucomicrobia门。
本研究结果表明, 有机背景土壤处理的细菌多样性高于常规背景的土壤处理, 并且加入秸秆的处理(OS)多样性最高。不管是在常规还是有机生产背景的土壤处理中, 加入苜蓿处理的多样性指数都要高于加入尿素的处理, 并且加入尿素后, 与对照相比多样性指数都有所下降, 表现出CA>C>CU, OA>O>OU的规律, 这与陈哲等[8]和徐永刚等[9]施入化肥导致细菌多样降低的研究结果一致, 这可能是因为相比于尿素, 苜蓿含有更多的营养元素, 可以更好的满足细菌的生长, 从而使得加入苜蓿的处理OA和CA增加了细菌的多样性。也可能是因为使用尿素后, 由于尿素过高的碳氮比, 打破了土壤养分的平衡, 从而影响了细菌的生长, 许多研究表明[10-12]土壤碳氮比是影响土壤微生物群落结构变化的一个重要因子。也可能是由于加入的尿素使土壤的N
从UPGMA聚类分析中可以看出, 常规土壤和有机土壤被分为2大类, 从细菌DGGE指纹图谱也可以看出, 常规和有机生产背景的特征性条带有明显区别。不同肥料的施入也引起了细菌群落的变化, 加入苜蓿处理的常规背景土壤CA和有机背景土壤OA分别出现了特征性条带B13和B29。
经过DGGE分离条带测序, 以及系统发育树的构建, 从亲缘关系上看, 常规背景土壤加入苜蓿的处理CA出现的特征性条带B13与Bacillus属很相似, 相似度为94%。Abbasi[14]等研究结果表明, Bacillus属有生物防治的潜力, 可以防治根结线虫, 可能是由于增加了大部分逆境酶的活性, 并且结果表明Bacillus属的Bacillus coagulans是一种潜在的促进植物生长的细菌。系统发育树归类显示, 第1簇Proteobacteria门的有B16、B28、B29等11个条带, 其中条带B16共同存在于常规和有机生产背景的土壤, 条带B28和B29仅仅存在于有机生产背景的土壤, 其中B16与B29分类不清楚, 与条带B28最相似菌株归属于Pseudomonas假单胞菌属, 假单胞菌属在近20多年来被较多的研究, 被认为是最具生防潜力和应用价值的一类细菌, 已有研究证明假单胞菌属可用于防治水稻纹枯病[15], 也可用于园艺作物如番茄和黄瓜由腐霉和茄丝核菌引起的苗期猝倒病的防治[16], 同时还可以增加某些园艺作物比如甜菜和萝卜的产量[17]。条带B24与B34仅仅存在于有机背景土壤的处理, 归属于第2簇Actinobacteria放线菌门, 同时由表3可以看出条带B19、B39也与放线菌门的相似性菌株相似度最高, 已有报道指出某些放线菌门细菌可以分解代谢某些除草剂[18], 同时也有很多报道指出放线菌可用于植物病害的生物防治[19]。
由以上讨论和DGGE图谱图1看出, B24、B28、B29、B34为有机生产背景处理土壤出现的条带, 条带B16、B19、B39虽然为共有条带, 但是有机背景土壤条带的亮度高于常规背景土壤, 已有研究指出在DGGE图谱中, 条带的亮度很可能代表微生物的相对丰度[20], 并且条带B13出现在经过苜蓿改良的常规背景土壤处理中, 因此有机农业或者是按照有机农业管理方式施肥的农田土壤更有益于防治植物病害, 有利于植物的生长。
The authors have declared that no competing interests exist.
| [1] |
Bacterial community structure and diversity in a century-old manure-treated agroecosystem [J].DOI:10.1128/AEM.70.10.5868-5874.2004 URL PMID: 15466526 [本文引用: 1] 摘要
Abstract Changes in soil microbial community structure and diversity may reflect environmental impact. We examined 16S rRNA gene fingerprints of bacterial communities in six agroecosystems by PCR amplification and denaturing gradient gel electrophoresis (PCR-DGGE) separation. These soils were treated with manure for over a century or different fertilizers for over 70 years. Bacterial community structure and diversity were affected by soil management practices, as evidenced by changes in the PCR-DGGE banding patterns. Bacterial community structure in the manure-treated soil was more closely related to the structure in the untreated soil than that in soils treated with inorganic fertilizers. Lime treatment had little effect on bacterial community structure. Soils treated with P and N-P had bacterial community structures more closely related to each other than to those of soils given other treatments. Among the soils tested, a significantly higher number of bacterial ribotypes and a more even distribution of the bacterial community existed in the manure-treated soil. Of the 99 clones obtained from the soil treated with manure for over a century, two (both Pseudomonas spp.) exhibited 100% similarity to sequences in the GenBank database. Two of the clones were possible chimeras. Based on similarity matching, the remaining 97 clones formed six major clusters. Fifty-six out of 97 were assigned taxonomic units which grouped into five major taxa: alpha-, beta-, and gamma-Proteobacteria (36 clones), Acidobacteria (16 clones), Bacteroidetes (2 clones), Nitrospirae (1 clone), and Firmicutes (1 clone). Forty-one clones remained unclassified. Results from this study suggested that bacterial community structure was closely related to agroecosystem management practices conducted for over 70 years.
|
| [2] |
Bacterial community structure and diversity in a black soil as affected by long-term fertilization [J].DOI:10.1016/S1002-0160(08)60052-1 Magsci [本文引用: 1] 摘要
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">Black soil (Mollisol) is one of the main soil types in northeastern China. Biolog and polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) methods were used to examine the influence of various fertilizer combinations on the structure and function of the bacterial community in a black soil collected from Harbin, Heilongjiang Province. Biolog results showed that substrate richness and catabolic diversity of the soil bacterial community were the greatest in the chemical fertilizer and chemical fertilizer+manure treatments. The metabolic ability of the bacterial community in the manure treatment was similar to the control. DGGE fingerprinting indicated similarity in the distribution of most 16S rDNA bands among all treatments, suggesting that microorganisms with those bands were stable and not influenced by fertilization. However, chemical fertilizer increased the diversity of soil bacterial community. Principal component analysis of Biolog and DGGE data revealed that the structure and function of the bacterial community were similar in the control and manure treatments, suggesting that the application of manure increased the soil microbial population, but had no effect on the bacterial community structure. Catabolic function was similar in the chemical fertilizer and chemical fertilizer+manure treatments, but the composition structure of the soil microbes differed between them. The use of chemical fertilizers could result in a decline in the catabolic activity of fast-growing or eutrophic bacteria.</p>
|
| [3] |
施肥和土壤管理对土壤微生物生物量碳、氮和群落结构的影响 [J].
以中国科学院沈阳生态试验站的长期定位试验为平台,研究了不同施肥和土壤管理对潮棕壤微生物生物量碳、氮和群落结构的影响。结果表明,裸地和农田处理的微生物生物量碳、氮较低,但是农田处理下施肥增加了微生物生物量,其中NPK+M效果最明显。DGGE图谱显示,处理间细菌条带分布较相似,其中裸地的细菌多样性最高;长期施肥和土壤管理改变了土壤真菌群落结构,施肥增加了真菌多样性,且有机肥的影响大于化肥;不同处理间氨氧化细菌群落结构差异显著,NPK+M显著增加了氨氧化细菌多样性,且无机肥和有机肥对氨氧化细菌群落影响不同。施肥和土壤管理对细菌影响较小,但显著改变了真菌和氨氧化细菌的群落结构。聚类分析结果显示,土壤管理措施较施肥对细菌、真菌和氨氧化细菌群落的影响更为显著。
|
| [4] |
Soil microbial biomass, dehydrogenase activity, bacterial community structure in response to long-term fertilizer management [J].DOI:10.1016/j.soilbio.2007.05.031 URL [本文引用: 1] 摘要
This study describes the effects of balanced versus nutrient-deficiency fertilization on soil microbial biomass, activity, and bacterial community structure in a long-term (16 years) field experiment. Long-term fertilization greatly increased soil microbial biomass C and dehydrogenase activity, except that the P-deficiency fertilization had no significant effect. Organic manure had a significantly greater ( P <0.05) impact on the biomass C and the activity, compared with mineral fertilizers. Microbial metabolic activity (dehydrogenase activity per microbial biomass C) was significantly higher ( P <0.05) under balanced fertilization than under nutrient-deficiency fertilization. General bacterial community structure was analyzed by PCR-denaturing gradient gel electrophoresis (DGGE) targeting eubacterial 16S rRNA gene. Mineral fertilization did not affect the DGGE banding pattern, while specific DGGE band was observed in organic manure-fertilized soils. Phylogenetic analysis showed that the change of bacterial community in organic manure-fertilized soil might not be because of the direct influence of the bacteria in the compost, but because of the promoting effect of the compost on the growth of an indigenous Bacillus sp. in the soil. We emphasize the importance of balanced-fertilization, as well as the role of P, in maintaining soil organic matter, and promoting the biomass and activity of microorganisms.
|
| [5] |
秸秆、有机肥及氮肥配合使用对水稻土微生物和有机质含量的影响 [J].
通过大田试验,研究了秸秆还田、施用有机肥和氮肥对水稻土微生物和土壤有机质含量的影响。试验结果表明:(1)在秸秆还田的情况下,增施氮肥(0-330 kg/hm2范围内)可促进秸秆的的腐解;(2)秸秆还田、施用有机肥和氮肥中的单一措施均能不同程度地增加水稻土壤细菌、真菌、放线菌的数量;(3)综合运用秸秆还田、施用有机肥和氮肥措施能协同增加土壤微生物数量,提高土壤生物量态氮和有机质含量,以秸秆还田量为6000 kg/hm2、施用有机肥量为4500 kg/hm2、施用氮肥量为240 kg/hm2效果较佳。
|
| [6] |
不同16SrDNA靶序列对DGGE分析活性污泥群落的影响 [J].DOI:10.3321/j.issn:0250-3301.2006.07.033 URL [本文引用: 1] 摘要
为探讨不同通用引物扩增16S rDNA靶序列对活性污泥微生物群落分析的影响,更合理的利用变性梯度凝胶电泳(DGGE)技术分析活性污泥样品.从连续流搅拌槽式反应器(CSTR)中 获取活性污泥,以3对通用引物341f/534r、968f/1 401r和341f/926r扩增16S rDNA序列,用DGGE分离PCR扩增产物.研究表明采用不同引物对进行DGGE分析时,群落多样性和动态存在显著的差异.341f/534r和 968f/1 401r的靶序列分离效果较好,341f/926r的靶序列分离效果较差.引物341f/534r和341f/926r DGGE图谱显示S2和S3相似性高,引物968f/1 401r DGGE图谱显示S1和S2相似性高.由此可见采用不同引物对进行DGGE分析时,群落结构之间的相似性和动态是不一致的.341f/534r的DGGE 图谱中条带丰富,多样性最好,968f/1 401r的DGGE图谱次之,341f/926r DGGE图谱条带最少,多样性也较差.因此,在利用DGGE分析活性污泥样品时采用引物341f/534r和968f/1 401r是比较适宜的.
|
| [7] |
Cultivable bacterial community from South China Sea sponge as revealed by DGGE fingerprinting and 16S rDNA phylogenetic analysis [J].DOI:10.1007/s00284-007-9035-2 Magsci [本文引用: 1] 摘要
<a name="Abs1"></a>The cultivable bacterial communities associated with four South China Sea sponges—<i>Stelletta tenuis</i>, <i>Halichondria rugosa</i>, <i>Dysidea avara</i>, and <i>Craniella australiensis</i> in mixed cultures—were investigated by microbial community DNA-based DGGE fingerprinting and 16S rDNA phylogenetic analysis. Diverse bacteria such as α-, γ-, δ-<i>Proteobacteria</i>, <i>Bacteroidetes</i>, and <i>Firmicutes</i> were cultured, some of which were previously uncultivable bacteria, potential novel strains with less than 95% similarity to their closest relatives and sponge symbionts growing only in the medium with the addition of sponge extract. According to 16S rDNA BLAST analysis, most of the bacteria were cultured from sponge for the first time, although similar phyla of bacteria have been previously recognized. The selective pressure of sponge extract on the cultured bacterial species was suggested, although the effect of sponge extract on bacterial community in high nutrient medium is not significant. Although α- and γ-<i>Proteobacteria</i> appeared to form the majority of the dominant cultivable bacterial communities of the four sponges, the composition of the cultivable bacterial community in the mixed culture was different, depending on the medium and sponge species. Greater bacterial diversity was observed in media C and CS for <i>Stelletta tenuis</i>, in media F and FS for <i>Halichondria rugosa</i> and <i>Craniella australiensis</i>. <i>S. tenuis</i> was found to have the highest cultivable bacterial diversity including α-, γ-, δ-<i>Proteobacteria</i>, <i>Bacteroidetes</i>, and <i>Firmicutes</i>, followed by sponge <i>Dysidea avara</i> without δ-<i>Proteobacteria</i>, sponge <i>Halichondria rugosa</i> with only α-, γ-<i>Proteobacteria</i> and <i>Bacteroidetes</i>, and sponge <i>C. australiensis</i> with only α-, γ-<i>Proteobacteria</i> and <i>Firmicutes</i>. Based on this study, by the strategy of mixed cultivation integrated with microbial community DNA-based DGGE fingerprinting and phylogenetic analysis, the cultivable bacterial community of sponge could be revealed effectively.
|
| [8] |
化肥对稻田土壤细菌多样性及硝化、反硝化功能菌组成的影响 [J].
以中国科学院桃源农业生态试验站长期定位施肥试验为平台,采用聚合酶链式反应(polymerase chain reaction, PCR)和DNA序列测定技术分析研究了3种长期施肥制度(对照不施肥-CK,单施氮肥-N,氮磷钾肥-NPK)对土壤细菌群落以及硝化、反硝化微生物种群的影响。通过系统分析细菌16S rDNA、细菌的硝化基因氨单加氧酶(ammonia monooxygenase, amoA)和反硝化基因氧化亚氮还原酶(nitrous oxide reductase, nosZ)等基因文库发现,长期单施氮肥导致细菌16S rDNA和amoA的多样性明显低于CK和NPK处理,而nosZ的多样性与之相反,即单施氮肥处理明显高于CK和NPK处理。LUBSHUFF软件统计分析显示:16S rDNA和amoA基因文库在CK与N,CK与NPK,NPK与N处理间均存在显著性差异。而对于nosZ基因文库,N和NPK与CK处理相比呈现出了显著性差异,N与NPK之间的差异没有达到显著水平。上述结果表明长期施用化肥对水稻土细菌的群落结构及硝化和反硝化细菌组成产生了明显的影响,但这种影响因基因类型而异。
|
| [9] |
长期不同施肥制度对潮棕壤微生物生物量碳、氮及细菌群落结构的影响 [J].
<p>以沈阳生态站长期定位试验为研究平台,采用传统氯仿熏蒸方法和现代PCR-DGGE技术探讨了长期不同施肥制度对土壤微生物生物量碳和氮及细菌群落结构的影响.结果表明:在整个试验期,土壤微生物生物量碳和氮的动态变化趋势基本相同;长期施用有机肥可显著提高土壤有机碳和土壤微生物生物量碳和氮含量,而长期施用化肥明显降低土壤pH,土壤微生物生物量碳和氮含量也显著降低.DGGE图谱表明:不同施肥处理的细菌16S rDNA多数条带分布相同,28条带中有18条为共有条带,说明潮棕壤中细菌类群较稳定,但其数量受到施肥的影响;长期施用有机肥促进潮棕壤细菌群落结构的多样性,而施用化肥处理则降低了其多样性.</p>
|
| [10] |
Effect of nitrogen and phosphate fertilisers on microbial and nematode diversity in pasture soils [J].DOI:10.1016/S0038-0717(00)00245-5 URL [本文引用: 1] 摘要
Soil microbial and nematode populations, soil microbial community structure, and microbial and nematode functional diversity were studied in two fertiliser trials on perennial pasture at three sampling times. The N trial involved the application of 0, 200 and 400 kg N ha(-1) y(-1) in the form of urea. The P trial involved the application of 0, 30, 50 and 100 kg P ha(-1) y(-1) as superphosphate. The purpose of this study was to determine biological characteristics that may be used as indicators of soil quality as affected by fertiliser inputs. The N or P treatments had no effect on total bacteria, cellulolytic microbes, or the fluorescein diacetate hydrolysis. The fungus Fusarium culmorum was found only in the 200 kg N treatment (P < 0.01). Gliocladium roseum declined in isolation frequency with increasing N (P < 0.05,) while other Gliocladium spp. increased (P < 0.01). The microbial community structure, ecophysiological index (EP), and colony-development index (CD) were determined using: colony development rates in 1/10 tryptose soy agar (TSA), a Pseudomonas medium, and a nutrient poor medium. These parameters were not affected by the addition of the N or P fertilisers. In the N trial, the functional diversity of soil microbes, as determined by Shannon Diversity Index (H) and average well colour development (AWCD) (using Biolog gram negative microplates) was higher in the unfertilised than fertilised treatments. The values for H and AWCD were 4.2 and 0.78 in the unfertilised compared to 4.0 and 0.53 in fertilised treatment (P < 0.01, 48 h, mean for both N treatments), respectively. There were no significant differences in these values in the P trial. Populations of the plant feeding nematodes Pratylenchus and Paratylenchus were greater (P < 0.05) whereas those of Meloidogyne were lower (P < 0.001), in soils fertilised with N than in unfertilised soils. The genera Aporcelaimus, Dorylaimellus, and Tylencholaimellus were found only in control plots and their loss paralleled faunal changes resulting from pasture improvement reported elsewhere. Nematode Maturity Index (MI) values were 1.78, 1.85, and 1.53 for the N fertiliser treatments (P < 0.05) suggesting a reduction at 400 kg N. The MI was not affected by the application of P (mean, 2.01), however, but all values in the P trial were greater than in the N trial. In the N and P trials an average of 29 and 35 nematode taxa were discriminated. The ratio of bacterial-feeding nematodes to bacterial-feeding plus fungal-feeding nematodes was similar across all treatments of the N (0.90-0.92) and P (0.84-0.90) trials, suggesting no relative change in the importance of bacterial- and fungal-mediated decomposition pathways in these soils as a result of fertiliser application. The finding that most microbiological characteristics did not respond to many years of fertiliser treatments suggests that the microbial community in the soils are similar and fertiliser amendments are insufficient to induce changes (either direct or indirect due to plant effects) in these communities. However, the consistent decrease in functional diversity of soil microflora and nematode populations with the application N, but not P, indicates that the N application can impact on community structure.
|
| [11] |
Orchard floor management practices that maintain vegetative or biomass groundcover stimulate soil microbial activity and alter soil microbial community composition [J].DOI:10.1007/s11104-004-3610-0 Magsci 摘要
<a name="Abs1"></a>Groundcover management systems (GMS) are important in managing fruit-tree orchards because of their effects on soil conditions, nutrient availability, tree growth and yields. We employed a polyphasic approach, incorporating measures of soil microbial abundance, activity and community composition, to study the long-term effects of different GMS on biotic and abiotic factors in an orchard soil. Four GMS treatments – Pre-emergence residual herbicides (Pre-H), post-emergence herbicide (Post-H), mowed-sod (Grass), and hardwood bark mulch (Mulch) – were established in 2-m-wide strips within tree rows in an apple orchard in 1992, and have been maintained and monitored annually until the present. We have measured soil water and nutrient availability, tree growth, and yields annually from 1993 to 2003. Soil nematode numbers and trophic groups were evaluated in July and Oct. 2001, and Sept. 2003. Numbers of culturable bacteria and fungi, soil respiratory activity, eubacterial and fungal community composition were determined in May and Sept. 2003. The Pre-H treatment soil had the fewest culturable bacteria, while the Grass treatment had the largest population of culturable fungi. Soil nematode population size and diversity were also affected by GMS treatments; the Pre-H treatment had the lowest ratio of (bacteriovores + fungivores) to plant parasitic nematodes. Soil respiration rates were higher in the Mulch than in other treatments during a 40-day incubation period. Hierarchical cluster dendrograms of denaturing gradient gel electrophoresis (DGGE) fingerprints for eubacterial community 16S rRNA genes indicated that Post-H and Grass treatments clustered together and separately from the Pre-H and Mulch treatments, which were also grouped together. The influence of GMSs on the fungal community, as assessed by PCR-DGGE of the internal transcribed spacer (ITS) region, was not as pronounced as that observed for bacteria. Soil fungal community composition under the Mulch differed from that under other treatments. The effects of GMS on soil microbial community abundance, activity, and composition were associated with observed differences in soil organic matter inputs and turnover, nutrient availability, and apple tree growth and yields under the different GMS treatments.
|
| [12] |
杉木人工林土壤质量演变过程中土壤微生物群落结构变化 [J].DOI:10.5846/stxb201205020628 Magsci [本文引用: 1] 摘要
以中国科学院会同森林生态站自然林、杉木连栽林和杉木阔叶树混交林地土壤为研究对象,采用PCR-DGGE、DNA-sequencing和主成分分析(PCA)等方法分析土壤微生物群落结构变化及其与土壤质量变化之间的关系。结果表明,该地区杉木人工林土壤中细菌优势种群为α-、β-、γ-proteobacteria和Cytophaga-Flexibacter-Bacterioides(CFB)类群;真菌优势种群为子囊菌(Ascomycetes)和担子菌(Basidiomycetes)亚门的种属。杉木人工林替代自然林后,土壤细菌多样性指数显著降低,且随连栽代数增加呈持续降低趋势;杉木人工林土壤中与<em>Pedobacter cryoconitis</em>亲缘关系密切的细菌种群消失,出现与<em>Xanthomonas</em> sp.和<em>Rhodanobacter</em> sp.亲缘关系密切的细菌种群。土壤真菌群落结构的变化与细菌相反,杉木人工林替代自然林后并不断连栽时,土壤真菌多样性指数呈现上升的趋势,自然林土壤中优势真菌种群在杉木三代林中消失。杉木与火力楠或桤木混交后土壤细菌和真菌种群结构与自然林类似。土壤细菌多样性指数与土壤总有机碳、全氮、可溶性有机碳、铵态氮、速效磷、速效钾含量以及土壤pH值呈显著正相关关系(<em>P</em><0.05)。土壤真菌多样性指数仅与土壤碳氮比呈显著正相关关系,而与土壤pH值呈显著负相关关系。杉木林土壤质量变化对土壤细菌和真菌优势种群有较大影响,细菌<em>Burkholderia</em> sp.、<em>Pedobacter</em> sp.、<em>Xanthomonas</em> sp.和真菌<em>Sclerotinia sclerotiorum</em>、<em>Mycosphaerella cannabis</em>可能是引起土壤质量变化的关键种群。
|
| [13] |
尿素肥斑扩散对土壤微生物群落结构的影响 [J].
磷脂脂肪酸 (PL FA)被认为是有效指示活体土壤微生物群落结构变化的标记物之一 ,该方法已在土壤微生物的研究中被大量应用。采用特制容器 ,模拟尿素肥斑在土壤中的扩散行为 ,观察距尿素肥斑不同距离微域中养分形态和浓度变化及其对土壤微生物群落结构的影响。试验结果表明 ,培养 7d后 ,No.7(距肥斑 7cm)和 No.8(距肥斑 8cm )微域的 NH+ 4、NO- 2 、NO- 3浓度最高 ,NO- 2 是尿素扩散区域的主要离子存在形态。对提取到的 2 5种 PL FA进行主成分分析 (PCA) ,发现 PL FA组成随不同微域养分浓度变化而变化 ,说明微生物群落结构发生了改变。就标记性 PL FA而言 ,尿素扩散导致真菌 PL FA在高浓度养分微域浓度增加 ,细菌 PL FA浓度下降 ,其中 ,No.7微域的真菌 PL FA18∶ 2ω6 ,9和 PL FA18∶ 1ω9浓度分别比对照 (No.2 0微域 )增加 173%和 4 7.2 %。然而 ,放线菌 PL FA 10 Me18∶ 0浓度变化不大。
|
| [14] |
Potential of Bacillus species against Meloidogyne javanica parasitizing eggplant(Solanum melongena L.)and induced biochemical changes [J].DOI:10.1007/s11104-013-1931-6 URL [本文引用: 1] 摘要
Aims The biocontrol potential of three Bacillus species, namely Bacillus subtilis (BS), Bacillus firmus (BF), and Bacillus coagulans (BC) was tested against the root-knot nematode Meloidogyne javanica (Treub) Chitwood in eggplants ( Solanum melongena L.). Plant growth and biochemical effects were also measured in these interactions. Methods Bacillus species were inoculated in soil around the seedlings of eggplants ( Solanum melongena L.) with and without nematodes in a greenhouse experiment. Plant growth, biochemical changes, and nematode parasitism were observed at 15 and 4502days after inoculation (DAI). Results BC significantly enhanced plant growth, chlorophyll “b” and total chlorophyll contents, and polyphenol oxidase (PPO) activity in the leaves of eggplants, while BS showed greatest reduction in root-knot nematode parasitism. Non-infected and untreated control (C61) plants showed lesser chlorophyll “b,” carotenoids, soluble protein contents, and guaiacol peroxidase but higher catalase and PPO activities compared to infected and untreated controls (C+) at 15 and 45 DAI. Superoxide dismutase activity declined in most of the treated plants at 45 DAI following rise at 15 DAI. Ascorbate peroxidase activity increased at 45 DAI compared to 15 DAI in C61 and C+ plants. PAL activity was greatly enhanced at 45 DAI in all treatments and controls over that at 15 DAI. Conclusions BC is a potentially plant growth-promoting bacteria although it was less effective against nematode infection compared to BS. Enzymes activities varied with infection and DAI. BC at 15 DAI in general increased the activity of most of the stress enzymes and thereby overcoming the effect of nematode parasitism.
|
| [15] |
铜绿假单胞菌ZJ1999对水稻纹枯病的防治及其在水稻上的定殖 [J].DOI:10.3321/j.issn:1005-9261.2006.01.013 URL [本文引用: 1] 摘要
利用拮抗菌铜绿假单胞菌 ZJ1999产生的粗提液进行防治水稻纹枯病研究的结果表明,抑制病菌侵染的活力随着粗提液浓度提高和处理时间的延长而增强。该菌株的定殖时间与最初引进 时的浓度密切相关,其最低应用浓度为108cfu/ml,在接种纹枯病病菌后第一天的喷施防效最佳。拮抗菌ZJ1999在发病的秧苗、成株期植株茎杆和叶 片上,14d后的菌量分别维持在102~103和103~104cfu/g。
|
| [16] |
铜绿假单胞菌株CR56在黄瓜和番茄根围的定殖能力 [J].DOI:10.3321/j.issn:1008-9209.2001.02.014 URL [本文引用: 1] 摘要
铜绿假单胞菌株 PseudomonasaeruginosaCR56作为种子处理时,能有效地防治由腐霉Pythiumspp.和茄丝核菌Rhizoctonia solania引起的黄瓜和番茄的苗期猝倒病,利用该菌的抗利福平突变菌株CR56R9和lacZY标记菌株CR56RL4,研究其在黄瓜和番茄根围的定 殖能力、种群动态和在根区的分布。研究表明,黄瓜根围比番茄根围更适合于该菌的定殖,其种群数量下降的幅度在番茄根围比在黄瓜根围更快。播后28d,菌株 CR56R9和CR56RL4在黄瓜根围种群数量分别由7.63×107CFU/粒和4.88×107CFU/粒下降到9.85×106CFU/株和 8.87×106CFU/株,在番茄根围则分别由3.0×107CFU/粒和2.13×107CFU/粒下降到4.48×105CFU/株和 4.37×105CFU/株;该菌主要分布在猝倒病的发生部位——根基部,在根尖处检测不到该菌的存在。lacZY标记菌株CR56RL4与抗利福平突变 菌株CR56R9,两者在黄瓜根围和番茄根围的存活能力基本一致。
|
| [17] |
Enhanced plant growth by siderophores produced by plant growth-promoting rhizobacteria [J].DOI:10.1038/286885a0 URL [本文引用: 1] 摘要
ABSTRACT Specific strains of the Pseudomonas fluorescens-putida group have recently been used as seed inoculants on crop plants to promote growth and increase yields. These pseudomonads, termed plant growth-promoting rhizobacteria (PGPR), rapidly colonize plant roots of potato, sugar beet and radish, and cause statistically significant yield increases up to 144% in field tests1-5. These results prompted us to investigate the mechanism by which plant growth was enhanced. A previous study indicated that PGPR increase plant growth by antagonism to potentially deleterious rhizoplane fungi and bacteria, but the nature of this antagonism was not determined6. We now present evidence that PGPR exert their plant growth-promoting activity by depriving native microflora of iron. PGPR produce extracellular siderophores (microbial iron transport agents)7 which efficiently complex environmental iron, making it less available to certain native microflora.
|
| [18] |
Characterization of s-triazine herbicide metabolism by a Nocardioides sp.isolated from agricultural soils [J].DOI:10.1128/AEM.66.8.3134-3141.2000 URL PMID: 10919761 [本文引用: 1] 摘要
Atrazine, a herbicide widely used in com production, is a frequently detected groundwater contaminant. Nine gram-positive bacterial strains able to use this herbicide as a sole source of nitrogen were isolated from four farms in central Canada. The strains were divided into two groups based on repetitive extragenic palindromic (rep)-PCR genomic fingerprinting with ERIC and BOXA1R primers. Based on 16S ribosomal DNA sequence analysis, both groups were identified as Nocardioides sp. strains. None of the isolates mineralized [ring-U-~(14)C]atrazine. There was no hybridization to genomic DNA from these strains using atzABC cloned from Pseudomonas sp. strain ADP or trzA cloned from Rhodococcus corallinus. S-Triazine degradation was studied in detail in Nocardioides sp. strain C190. Oxygen was not required for atrazine degradation by whole cells or cell extracts. Based on high-pressure liquid chromatography and mass spectrometric analyses of products formed from atrazine in incubations of whole cells with H_2~(18)O, sequential hydrolytic reactions converted atrazine to hydroxyatrazine and then to the end product N-ethylammelide. lsopropylamine, the putative product of the second hydrolytic reaction, supported growth as the sole carbon and nitrogen source. The triazine hydrolase from strain C190 was isolated and purified and found to have a K_m for atrazine of 25 #mu#M and a V_(max) of 31 btmol/min/mg of protein. The subunit molecular mass of the protein was 52 kDa. Atrazine hydrolysis was not inhibited by 500 #mu#m EDTA but was inhibited by 100 #mu#M G, Cu, Co, or Zn. Whole cells and purified triazine hydrolase converted a range of chlorine or methylthio.substituted herbicides to the corresponding hydroxy derivatives. In summary, an atrazine-metabolizing Nocardioides sp. widely distributed in agricultural soils degrades a range of s-triazine herbicides by means of a novel s.triazine hydrolase.
|
| [19] |
放线菌在植病生防中的研究进展 [J].DOI:10.3969/j.issn.2095-0896.2005.05.024 URL [本文引用: 1] 摘要
综述了植物病害生物防治研究的意义,详细介绍了放线菌在植物病害生物防治中的拮抗机制及其生防作用,并对一些有关放线菌应用中的问题及解决途径进行了探讨。
|
| [20] |
De Waal E C, Uitterlinden A G.Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA [J].DOI:10.1080/15389588.2015.1013535 URL PMID: 202176 [本文引用: 1] 摘要
We describe a new molecular approach to analyzing the genetic diversity of complex microbial populations. This technique is based on the separation of polymerase chain reaction-amplified fragments of genes coding for 16S rRNA, all the same length, by denaturing gradient gel electrophoresis (DGGE). DGGE analysis of different microbial communities demonstrated the presence of up to 10 distinguishable bands in the separation pattern, which were most likely derived from as many different species constituting these populations, and thereby generated a DGGE profile of the populations. We showed that it is possible to identify constituents which represent only 1% of the total population. With an oligonucleotide probe specific for the V3 region of 16S rRNA of sulfate-reducing bacteria, particular DNA fragments from some of the microbial populations could be identified by hybridization analysis. Analysis of the genomic DNA from a bacterial biofilm grown under aerobic conditions suggests that sulfate-reducing bacteria, despite their anaerobicity, were present in this environment. The results we obtained demonstrate that this technique will contribute to our understanding of the genetic diversity of uncharacterized microbial populations.
|

| 沪交ICP备05221 版权所有:《上海交通大学学报(农业科学版)》编辑部 主管单位:中华人民共和国教育部 主办单位:上海交通大学 出版单位:上海交通大学学报编辑部 地址:上海市七莘路2678号 上海交通大学七宝校区36号信箱 邮政编码:201101 电话:021-64789728 电子邮件:xuebao@sjtu.edu.cn |
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}