诊断学理论与实践 ›› 2025, Vol. 24 ›› Issue (05): 542-547.doi: 10.16150/j.1671-2870.2025.05.010
谢靓哲, 戴菁, 武文漫, 王学锋( )
)
收稿日期:2023-09-01
修回日期:2024-10-28
接受日期:2024-12-05
出版日期:2025-10-25
发布日期:2025-10-23
通讯作者:
王学锋 E-mail: wxf10339@rjh.com.cn基金资助:
XIE Liangzhe, DAI Jing, WU Wenman, WANG Xuefeng()
Received:2023-09-01
Revised:2024-10-28
Accepted:2024-12-05
Published:2025-10-25
Online:2025-10-23
摘要:
凝血因子Ⅷ(factor Ⅷ,FⅧ) 缺乏症是一种罕见的出血性疾病,可表现为自发性或延迟性危及生命的出血。FⅧ由2个催化亚基(FⅧ-A2)和2个载体亚基(FⅧ-B2)组成,除了在止血中发挥重要的作用外,还具有多种功能,包括血管生成、维持妊娠、伤口愈合和骨代谢等。FⅧ缺乏根据病因的不同分为遗传性和获得性两类,遗传性FⅧ缺乏症全球发病率为0.05/10万。大多数遗传性FⅧ缺乏患者通常表现为FⅧ-A缺乏。获得性FⅧ缺乏症通常由过度消耗和合成减少等因素引起,比遗传性更为常见。在极少数情况下,获得性FⅧ缺乏症患者可能会产生针对FⅧ亚基的抑制物,这可能是特发性的,也可能与合并症(如恶性肿瘤或自身免疫性疾病有关)。诊断FⅧ缺乏症的一线检测方法是FⅧ活性定量检测。对于遗传性FⅧ缺乏症患者,需要通过酶联免疫吸附试验测定其FⅧ缺乏的类型,并检测相关分子遗传突变。若考虑获得性FⅧ缺乏症,还需要通过免疫试验检测抑制物。FⅧ缺乏症治疗主要包括FⅧ替代治疗和(或)免疫抑制治疗。FⅧ替代选择已经从传统的新鲜冷冻血浆、旧血浆、全血和低温沉淀,发展到血浆衍生和重组FⅧ浓缩物。虽然FⅧ缺乏症的治疗靶点和阈值目前尚未明确,但对严重的FⅧ缺乏症患者进行及时诊断和适当管理,可以显著降低其发病率和死亡率。
中图分类号:
谢靓哲, 戴菁, 武文漫, 王学锋. 凝血因子Ⅷ缺乏症研究进展[J]. 诊断学理论与实践, 2025, 24(05): 542-547.
XIE Liangzhe, DAI Jing, WU Wenman, WANG Xuefeng. Research advances in coagulation factor Ⅷ deficiency[J]. Journal of Diagnostics Concepts & Practice, 2025, 24(05): 542-547.
表1
获得性FⅧ缺乏症的相关疾病、诊断和治疗特点
| 分类 | 病理生理学 | 相关疾病 | 诊断 | 治疗 |
|---|---|---|---|---|
| 自身免疫性 | 自身抗体介导的抑制或快速清除 | 自身免疫性疾病:系统性红斑狼疮、类风湿性关节炎 恶性肿瘤:实体瘤、血液系统疾病 药物:异烟肼等 意义未明的单克隆免疫球蛋白血症等 | 临床表现:严重出血 实验室检查:FⅧ活性<10%,Bethesda试验(中和抗体),结合试验(非中和抗体) | 止血治疗:大剂量FⅧ、抗纤溶 |
| 非自身免疫性 | 过度消耗或 合成减少 | 手术、弥散性血管内凝血、炎症性肠病、过敏性紫癜、脓毒血症、血栓形成、肺栓塞、卒中等 肝病、白血病 药物:丙戊酸钠、托珠单抗等 | 临床表现:出血不严重 实验室检查:FⅧ活性20%~70%,其他凝血因子可能缺乏 | +抗体消除:免疫抑制剂、血浆置换 |
表2
ISTH对自身抗体介导的FⅧ缺乏的诊断标准的建议
| 自身免疫性FⅧ缺乏症的诊断标准 |
|---|
| 可能: |
| 1、近期出现出血症状,主要发生在老年人 |
| 2、无FⅧ或其他凝血因子先天性/遗传性缺陷家族史 |
| 3、既往无出血症状,特别是与止血困难相关事件(如手术、侵入性检查、创伤等) |
| 4、与抗凝药和抗血小板药物等无关 |
| 5、实验室检测FⅧ参数异常[FⅧ活性和(或)抗原< 50%] |
| 可信: |
| 第1-5项加上存在FⅧ抑制剂(37 ℃培养2 h后,患者和健康对照组血浆交叉混合试验呈阳性) |
| 确诊: |
| 第1-5项加上抗FⅧ自身抗体的存在(免疫方法阳性) |
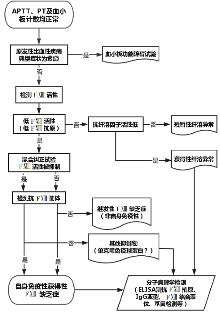
图1
获得性FⅧ缺乏症伴出血症状的诊断流程图[10]
| [1] | YAN M T S, RYDZ N, GOODYEAR D, et al. Acquired factor Ⅷ deficiency: A review[J]. Transfus Apher Sci, 2018, 57(6):724-730. |
| [2] | NADERI M, DORGALALEH A, TABIBIAN SH, et al. Current understanding in diagnosis and management of factor Ⅷ deficiency[J]. Iran J Ped Hematol Oncol, 2013, 3(4):164-172. |
| [3] | MUSZBEK L, BERECZKY Z, BAGOLY Z, et al. Factor Ⅷ: a coagulation factor with multiple plasmatic and cellular functions[J]. Physiol Rev, 2011, 91(3):931-972. |
| [4] | THOMAS V, EL ALAOUI S, MASSIGNON D, et al. Development and evaluation of a modified colorimetric solid-phase microassay for measuring the activity of cellular and plasma (factor Ⅷ) transglutaminases[J]. Biotechnol Appl Biochem, 2006, 43(Pt 3):171-179. |
| [5] | KLEBER C, SABLOTZKI A, CASU S, et al. The impact of acquired coagulation factor Ⅷ deficiency in traumatic bleeding and wound healing[J]. Crit Care, 2022, 26(1):69. |
| [6] | DORGALALEH A, NADERI M, HOSSEINI M S, et al. factor Ⅷ deficiency in Iran: a comprehensive review of the literature[J]. Semin Thromb Hemost, 2015, 41(3):323-329. |
| [7] | DORGALALEH A, TABIBIAN S, HOSSEINI M S, et al. Diagnosis of factor Ⅷ deficiency[J]. Hematology, 2016, 21(7):430-439. |
| [8] | LEVY J H, GREENBERG C. Biology of factor Ⅷ and clinical manifestations of factor Ⅷ deficiency[J]. Transfusion, 2013, 53(5):1120-1131. |
| [9] | PELCOVITS A, SCHIFFMAN F, NIROULA R. Factor Ⅷ deficiency: A review of clinical presentation and management[J]. Hematol Oncol Clin North Am, 2021, 35(6):1171-1180. |
| [10] | AMANO S, OKA K, SATO Y, et al. Measuring factor Ⅷ inhibitors in patients with factor Ⅷ deficiency: A case report and systematic review of current practices in Japan[J]. J Clin Med, 2022, 11(6):1699. |
| [11] |
PEYVANDI F, PALLA R, MENEGATTI M, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders[J]. J Thromb Haemost, 2012, 10(4):615-621.
doi: 10.1111/j.1538-7836.2012.04653.x pmid: 22321862 |
| [12] | KOSEKI-KUNO S, YAMAKAWA M, DICKNEITE G, et al. factor Ⅷ A subunit-deficient mice developed severe uterine bleeding events and subsequent spontaneous miscarriages[J]. Blood, 2003, 102(13):4410-4412. |
| [13] | ICHINOSE A, Japanese Collaborative Research Group on AH13. Autoimmune acquired factor Ⅷ deficiency due to anti-factor Ⅷ/13 antibodies: A summary of 93 patients[J]. Blood Rev, 2017, 31(1):37-45. |
| [14] | ICHINOSE A, KOHLER H P, PHILIPPOU H. Recommendation for ISTH/SSC Criterion 2015 for autoimmune acquired factor Ⅷ/13 deficiency[J]. Thromb Haemost, 2016, 116(4):772-774. |
| [15] | AHMAD F, SOLYMOSS S, POON M C, et al. Characte-rization of an acquired IgG inhibitor of coagulation factor Ⅷ in a patient with systemic lupus erythematosus[J]. Br J Haematol, 1996, 93(3):700-703. |
| [16] | LIM W, MOFFAT K, HAYWARD C P. Prophylactic and perioperative replacement therapy for acquired factor Ⅷ deficiency[J]. J Thromb Haemost, 2004, 2(6):1017-1019. |
| [17] | MUSZBEK L, PÉNZES K, KATONA É. Auto- and alloantibodies against factor Ⅷ: laboratory diagnosis and clinical consequences[J]. J Thromb Haemost, 2018, 16(5):822-832. |
| [18] | ICHINOSE A. Hemorrhagic acquired factor Ⅷ (13) deficiency and acquired hemorrhaphilia 13 revisited[J]. Semin Thromb Hemost, 2011, 37(4):382-388. |
| [19] | AJZNER E, SCHLAMMADINGER A, KERÉNYI A, et al. Severe bleeding complications caused by an autoantibody against the B subunit of plasma factor Ⅷ: a novel form of acquired factor Ⅷ deficiency[J]. Blood, 2009, 113(3):723-725. |
| [20] | SOURI M, OSAKI T, ICHINOSE A. Anti-factor Ⅷ A subunit (FⅧ-A) autoantibodies block FⅧ-A2 B2 assembly and steal FⅧ-A from native FⅧ-A2 B2[J]. J Thromb Haemost, 2015, 13(5):802-814. |
| [21] |
LUO Y Y, ZHANG G S. Acquired factor Ⅷ inhibitor: clinical features, treatment, fibrin structure and epitope determination[J]. Haemophilia, 2011, 17(3):393-398.
doi: 10.1111/j.1365-2516.2010.02459.x pmid: 21323797 |
| [22] | HAYASHI T, KADOHIRA Y, MORISHITA E, et al. A case of acquired FⅧ deficiency with severe bleeding symptoms[J]. Haemophilia, 2012, 18(4):618-620. |
| [23] | NIJENHUIS A V, VAN BERGEIJK L, HUIJGENS P C, et al. Acquired factor Ⅷ deficiency due to an inhibitor: a case report and review of the literature[J]. Haematologica, 2004, 89(5):ECR14. |
| [24] | KATONA É, MUSZBEK L. Diagnosis and management of congenital and acquired FⅧ deficiencies[J] Semin Thromb Hemost, 2016, 42(4):429-439. |
| [25] | KOHLER H P, ICHINOSE A, SEITZ R, et al. Factor Ⅷ and fibrinogen SSC subcommittee of the ISTH. Diagnosis and classification of factor Ⅷ deficiencies[J]. J Thromb Haemost, 2011, 9(7):1404-1406. |
| [26] | SCHROEDER V, KOHLER H P. Factor Ⅷ deficiency: an update[J]. Semin Thromb Hemost, 2013, 39(6):632-641. |
| [27] | BOVET J, HURJÁK B, DE MAISTRE E, et al. Autoimmune factor Ⅷ deficiency with unusual laboratory and clinical phenotype[J]. J Thromb Haemost, 2020, 18(6):1330-1334. |
| [28] | DORGALALEH A, KAZEMI A, ZAKER F, et al. Laboratory diagnosis of factor Ⅷ deficiency, routine coagulation tests with quantitative and qualitative methods[J]. Clin Lab, 2016, 62(4):491-498. |
| [29] | KARIMI M, PEYVANDI F, NADERI M, et al. Factor Ⅷ deficiency diagnosis: Challenges and tools[J]. Int J Lab Hematol, 2018, 40(1):3-11. |
| [30] | BOEHLEN F, CASINI A, CHIZZOLINI C, et al. Acquired factor Ⅷ deficiency: a therapeutic challenge[J]. Thromb Haemost, 2013, 109(3):479-487. |
| [31] | FRANCHINI M, FRATTINI F, CRESTANI S, et al. Acquired FⅧ inhibitors: a systematic review[J]. J Thromb Thrombolysis, 2013, 36(1):109-114. |
| [32] | TONE K J, JAMES T E, FERGUSSON D A, et al. Acquired factor Ⅷ inhibitor in hospitalized and periope-rative patients: A systematic review of case reports and case series[J]. Transfus Med Rev, 2016, 30(3):123-131. |
| [33] | MIESBACH W. Rituximab in the treatment of factor Ⅷ inhibitor possibly caused by Ciprofloxacin[J]. Thromb Haemost, 2005, 93(5):1001-1003. |
| [34] | NGO SACK F, GALINAT H, EGRETEAU P Y, et al. Efficacy of rituximab in acquired factor Ⅷ inhibitor after arterial rFVIIa-induced thrombosis[J]. Haemophilia, 2013, 19(2):e93-e94. |
| [35] | DREYFUS M, BARROIS D, BORG J Y, et al. Successful long-term replacement therapy with concentrate (Fibrogammin(®) P) for severe congenital factor Ⅷ deficiency: a prospective multicentre study[J]. J Thromb Haemost, 2011, 9(6):1264-1266. |
| [36] | LASSILA R. Clinical use of factor Ⅷ concentrates[J]. Semin Thromb Hemost, 2016, 42(4):440-444. |
| [37] | SEITZ R, DUCKERT F, LOPACIUK S, et al. ETRO Working Party on Factor Ⅷ questionnaire on congeniatal factor Ⅷ deficiency in Europe: status and perspectives. Study Group[J]. Semin Thromb Hemost, 1996, 22(5):415-418. |
| [38] | MAHMOODI M, PEYVANDI F, AFRASIABI A, et al. Bleeding symptoms in heterozygous carriers of inherited coagulation disorders in southern Iran[J]. Blood Coagul Fibrinolysis, 2011, 22(5):396-401. |
| [39] | JANBAIN M, NUGENT D J, POWELL J S, et al. Use of factor Ⅷ (FⅧ) concentrate in patients with congenital Ⅷ deficiency undergoing surgical procedures[J]. Transfusion, 2015, 55(1):45-50. |
| [40] | DORGALALEH A, RASHIDPANAH J. Blood coagulation factor Ⅷ and factor Ⅷ deficiency[J]. Blood Rev, 2016, 30(6):461-475. |
| [1] | 林莉亚, 吴希, 毛胤祺, 陈光明, 武文漫, 戴菁, 王学锋, 丁秋兰. 三种位于ADAMTS13编码去整合素样结构域上的基因突变可导致蛋白产物功能缺陷并又发血栓形成[J]. 诊断学理论与实践, 2025, 24(04): 431-440. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||