Background Alzheimer's disease (AD) is a neurodegenerative disorder and the predominant type of dementia worldwide. It is characterized by the progressive and irreversible decline of cognitive functions. In addition to the pathological beta-amyloid (Aβ) deposition, glial activation, and neuronal injury in the postmortem brains of AD patients, increasing evidence suggests that the often overlooked vascular dysfunction is an important early event in AD pathophysiology. Vascular endothelial growth factor (VEGF) plays a critical role in regulating physiological functions and pathological changes in blood vessels, but whether VEGF is involved in the early stage of vascular pathology in AD remains unclear.
Methods We used an antiangiogenic agent for clinical cancer treatment, the humanized monoclonal anti-VEGF antibody bevacizumab, to block VEGF binding to its receptors in the 5×FAD mouse model at an early age. After treatment, memory performance was evaluated by a novel object recognition test, and cerebral vascular permeability and perfusion were examined by an Evans blue assay and blood flow scanning imaging analysis. Immunofluorescence staining was used to measure glial activation and Aβ deposits. VEGF and its receptors were analyzed by enzyme-linked immunosorbent assay and immunoblotting. RNA sequencing was performed to elucidate bevacizumab-associated transcriptional signatures in the hippocampus of 5×FAD mice.
Results Bevacizumab treatment administered from 4 months of age dramatically improved cerebrovascular functions, reduced glial activation, and restored long-term memory in both sexes of 5×FAD mice. Notably, a sex-specific change in different VEGF receptors was identified in the cortex and hippocampus of 5×FAD mice. Soluble VEGFR1 was decreased in female mice, while full-length VEGFR2 was increased in male mice. Bevacizumab treatment reversed the altered expression of receptors to be comparable to the level in the wild-type mice. Gene Set Enrichment Analysis of transcriptomic changes revealed that bevacizumab effectively reversed the changes in the gene sets associated with blood-brain barrier integrity and vascular smooth muscle contraction in 5×FAD mice.
Conclusions Our study demonstrated the mechanistic roles of VEGF at the early stage of amyloidopathy and the protective effects of bevacizumab on cerebrovascular function and memory performance in 5×FAD mice. These findings also suggest the therapeutic potential of bevacizumab for the early intervention of AD.
Background Alzheimer's disease (AD) is considered to have a multifactorial etiology. The hallmark of AD is progressive neurodegeneration, which is characterized by the deepening loss of memory and a high mortality rate in the elderly. The neurodegeneration in AD is believed to be exacerbated following the intercoupled cascades of extracellular amyloid beta (Aβ) plaques, uncontrolled microglial activation, and neuroinflammation. Current therapies for AD are mostly designed to target the symptoms, with limited ability to address the mechanistic triggers for the disease. In this study, we report a novel nanotechnology based on microglial scavenger receptor (SR)-targeting amphiphilic nanoparticles (NPs) for the convergent alleviation of fibril Aβ (fAβ) burden, microglial modulation, and neuroprotection.
Methods We designed a nanotechnology approach to regulate the SR-mediated intracellular fAβ trafficking within microglia. We synthesized SR-targeting sugar-based amphiphilic macromolecules (AM) and used them as a bioactive shell to fabricate serum-stable AM-NPs via flash nanoprecipitation. Using electron microscopy, in vitro approaches, ELISA, and confocal microscopy, we investigated the effect of AM-NPs on Aβ fibrilization, fAβ-mediated microglial inflammation, and neurotoxicity in BV2 microglia and SH-SY5Y neuroblastoma cell lines.
Results AM-NPs interrupted Aβ fibrilization, attenuated fAβ microglial internalization via targeting the fAβ-specific SRs, arrested the fAβ-mediated microglial activation and pro-inflammatory response, and accelerated lysosomal degradation of intracellular fAβ. Moreover, AM-NPs counteracted the microglial-mediated neurotoxicity after exposure to fAβ.
Conclusions The AM-NP nanotechnology presents a multifactorial strategy to target pathological Aβ aggregation and arrest the fAβ-mediated pathological progression in microglia and neurons.
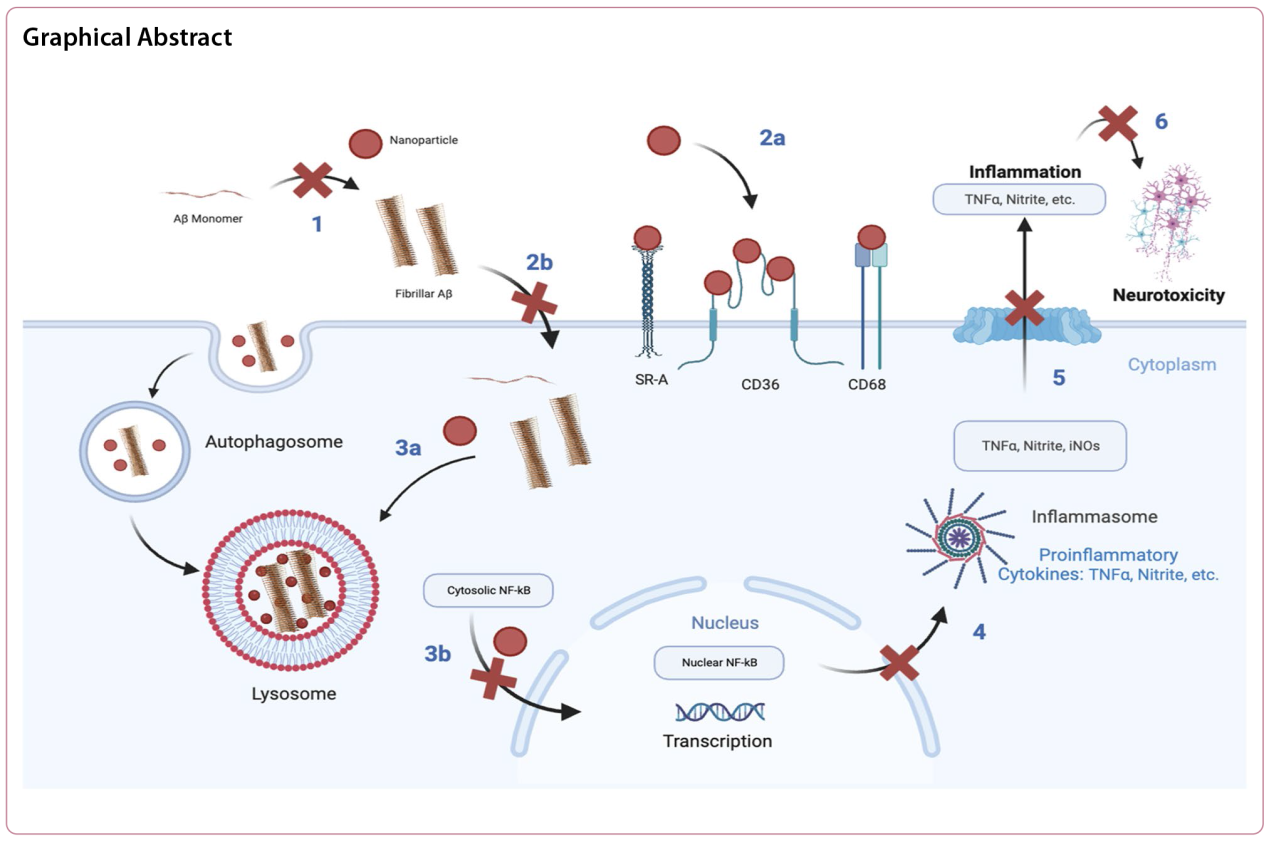
Background Microglia-mediated neuroinflammation in Alzheimer’s disease (AD) is not only a response to pathophysiological events, but also plays a causative role in neurodegeneration. Cytoplasmic cysteinyl-tRNA synthetase (CARS) is considered to be a stimulant for immune responses to diseases; however, it remains unknown whether CARS is involved in the pathogenesis of AD.
Methods Postmortem human temporal cortical tissues at different Braak stages and AD patient-derived serum samples were used to investigate the changes of CARS levels in AD by immunocytochemical staining, real-time PCR, western blotting and ELISA. After that, C57BL/6J and APP/PS1 transgenic mice and BV-2 cell line were used to explore the role of CARS protein in memory and neuroinflammation, as well as the underlying mechanisms. Finally, the associations of morphological features among CARS protein, microglia and dense-core plaques were examined by immunocytochemical staining.
Results A positive correlation was found between aging and the intensity of CARS immunoreactivity in the temporal cortex. Both protein and mRNA levels of CARS were increased in the temporal cortex of AD patients. Immunocytochemical staining revealed increased CARS immunoreactivity in neurons of the temporal cortex in AD patients. Moreover, overexpression of CARS in hippocampal neurons induced and aggravated cognitive dysfunction in C57BL/6J and APP/PS1 mice, respectively, accompanied by activation of microglia and the TLR2/MyD88 signaling pathway as well as upregulation of proinflammatory cytokines. In vitro experiments showed that CARS treatment facilitated the production of proinflammatory cytokines and the activation of the TLR2/MyD88 signaling pathway of BV-2 cells. The accumulation of CARS protein occurred within dense-core Aβ plaques accompanied by recruitment of ameboid microglia. Significant upregulation of TLR2/MyD88 proteins was also observed in the temporal cortex of AD.
Conclusions The findings suggest that the neuronal CARS drives neuroinflammation and induces memory deficits, which might be involved in the pathogenesis of AD.
Alzheimer’s disease (AD) is the most prevalent form of dementia in the elderly and represents a major clinical challenge in the ageing society. Neuropathological hallmarks of AD include neurofibrillary tangles composed of hyperphosphorylated tau, senile plaques derived from the deposition of amyloid-β (Aβ) peptides, brain atrophy induced by neuronal loss, and synaptic dysfunctions. Death-associated protein kinase 1 (DAPK1) is ubiquitously expressed in the central nervous system. Dysregulation of DAPK1 has been shown to contribute to various neurological diseases including AD, ischemic stroke and Parkinson’s disease (PD). We have established an upstream effect of DAPK1 on Aβ and tau pathologies and neuronal apoptosis through kinase-mediated protein phosphorylation, supporting a causal role of DAPK1 in the pathophysiology of AD. In this review, we summarize current knowledge about how DAPK1 is involved in various AD pathological changes including tau hyperphosphorylation, Aβ deposition, neuronal cell death and synaptic degeneration. The underlying molecular mechanisms of DAPK1 dysregulation in AD are discussed. We also review the recent progress regarding the development of novel DAPK1 modulators and their potential applications in AD intervention. These findings substantiate DAPK1 as a novel therapeutic target for the development of multifunctional disease-modifying treatments for AD and other neurological disorders.
Neurodegenerative disorders present complex pathologies characterized by various interconnected factors, including the aggregation of misfolded proteins, oxidative stress, neuroinflammation and compromised blood-brain barrier (BBB) integrity. Addressing such multifaceted pathways necessitates the development of multi-target therapeutic strategies. Emerging research indicates that probucol, a historic lipid-lowering medication, offers substantial potential in the realm of neurodegenerative disease prevention and treatment. Preclinical investigations have unveiled multifaceted cellular effects of probucol, showcasing its remarkable antioxidative and anti-inflammatory properties, its ability to fortify the BBB and its direct influence on neural preservation and adaptability. These diverse effects collectively translate into enhancements in both motor and cognitive functions. This review provides a comprehensive overview of recent findings highlighting the efficacy of probucol and probucol-related compounds in the context of various neurodegenerative conditions, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and cognitive impairment associated with diabetes.
Ageing is a crucial risk factor for Alzheimer’s disease (AD) and is characterised by systemic changes in both intracellular and extracellular microenvironments that affect the entire body instead of a single organ. Understanding the specific mechanisms underlying the role of ageing in disease development can facilitate the treatment of ageing-related diseases, such as AD. Signs of brain ageing have been observed in both AD patients and animal models. Alleviating the pathological changes caused by brain ageing can dramatically ameliorate the amyloid beta- and tau-induced neuropathological and memory impairments, indicating that ageing plays a crucial role in the pathophysiological process of AD. In this review, we summarize the impact of several age-related factors on AD and propose that preventing pathological changes caused by brain ageing is a promising strategy for improving cognitive health.
Background Little is known about the impact of the COVID-19 pandemic on patients with Parkinson’s disease (PD) at different stages of the pandemic. This study aims to assess the lives and disease status of PD patients during the zero-COVID policy period and after ending the zero-COVID policy.
Methods This multicenter cross-sectional study included two online surveys among PD patients in China, from May 30 to June 30 in 2022 and from January 1 to February 28 in 2023, respectively. The survey questionnaires contained four sections: (1) status of COVID-19 infection; (2) impact on motor and non-motor symptoms; (3) impact on daily and social lives; and (4) impact on PD disease management.
Results A total of 1764 PD patients participated in the first online survey, with 200 patients having lockdown experience and 3 being COVID-19-positive (0.17%). In addition, 537 patients participated in the second online survey, with 467 patients having COVID-19 infection (86.96%). (1) During zero-COVID, all of the COVID-19-positive patients had mild symptoms of COVID-19 and no death was reported. After zero-COVID, 83.51% of the COVID-19-positive patients had mild symptoms. The overall death rate and inpatient mortality rate of COVID-19-positive PD patients were 3.21% and 30.00%, respectively. (2) During zero-COVID, 49.43% of PD patients reported worsening of PD-related symptoms (lockdown vs. unlockdown, 60.50% vs. 48.02%, P = 0.0009). After zero-COVID, 54.93% of PD patients reported worsening of PD-related symptoms (COVID-19 positive vs. COVID-19 negative, 59.31% vs. 25.71%, P < 0.0001). (3) During zero-COVID, 62.36% of patients felt worried, and ‘limited outdoor activities’ (55.39%) was the top reason for mental health problems. After zero-COVID, 59.03% of patients felt worried, with ‘poor health’ (58.10%) being the top reason. The PD patients tended to change their daily activities from offline to online, and their economic and caregiver burdens increased both during and after zero-COVID. (4) Most PD patients would like to choose online rehabilitation during (69.56%) and after zero-COVID (69.27%). The demand for online medication purchasing also increased during (47.00%) and after zero-COVID (26.63%).
Conclusions The COVID-19 pandemic aggravated the motor and non-motor symptoms of PD patients either during or after the zero-COVID policy period. The PD patients also experienced prominent mental health problems, changes in daily activities, and increases in economic and caregiver burdens. The COVID-19 pandemic has changed ways of PD management with increasing demands for online medication purchasing and rehabilitation.
Background Degeneration of the locus coeruleus (LC) noradrenergic system contributes to clinical symptoms in Alzheimer’s disease (AD) and Parkinson’s disease (PD). Diffusion magnetic resonance imaging (MRI) has the potential to evaluate the integrity of the LC noradrenergic system. The aim of the current study was to determine whether the diffusion MRI-measured integrity of the LC and its tracts are sensitive to noradrenergic degeneration in AD and PD.
Methods Post-mortem in situ T1-weighted and multi-shell diffusion MRI was performed for 9 AD, 14 PD, and 8 control brain donors. Fractional anisotropy (FA) and mean diffusivity were derived from the LC, and from tracts between the LC and the anterior cingulate cortex, the dorsolateral prefrontal cortex (DLPFC), the primary motor cortex (M1) or the hippocampus. Brain tissue sections of the LC and cortical regions were obtained and immunostained for dopamine-beta hydroxylase (DBH) to quantify noradrenergic cell density and fiber load. Group comparisons and correlations between outcome measures were performed using linear regression and partial correlations.
Results The AD and PD cases showed loss of LC noradrenergic cells and fibers. In the cortex, the AD cases showed increased DBH + immunoreactivity in the DLPFC compared to PD cases and controls, while PD cases showed reduced DBH + immunoreactivity in the M1 compared to controls. Higher FA within the LC was found for AD, which was correlated with loss of noradrenergic cells and fibers in the LC. Increased FA of the LC-DLPFC tract was correlated with LC noradrenergic fiber loss in the combined AD and control group, whereas the increased FA of the LC-M1 tract was correlated with LC noradrenergic neuronal loss in the combined PD and control group. The tract alterations were not correlated with cortical DBH + immunoreactivity.
Conclusions In AD and PD, the diffusion MRI-detected alterations within the LC and its tracts to the DLPFC and the M1 were associated with local noradrenergic neuronal loss within the LC, rather than noradrenergic changes in the cortex.
Brain aging is a recognized risk factor for neurodegenerative diseases like Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS, Lou Gehrig's disease), but the intricate interplay between brain aging and the pathogenesis of these conditions remains inadequately understood. Cellular senescence is considered to contribute to cellular dysfunction and inflammaging. According to the threshold theory of senescent cell accumulation, the vulnerability to neurodegenerative diseases is associated with the rates of senescent cell generation and clearance within the brain. Given the role of microglia in eliminating senescent cells, the accumulation of senescent microglia may lead to the acceleration of brain aging, contributing to inflammaging and increased vulnerability to neurodegenerative diseases. In this review, we propose the idea that the senescence of microglia, which is notably vulnerable to aging, could potentially serve as a central catalyst in the progression of neurodegenerative diseases. The senescent microglia are emerging as a promising target for mitigating neurodegenerative diseases.
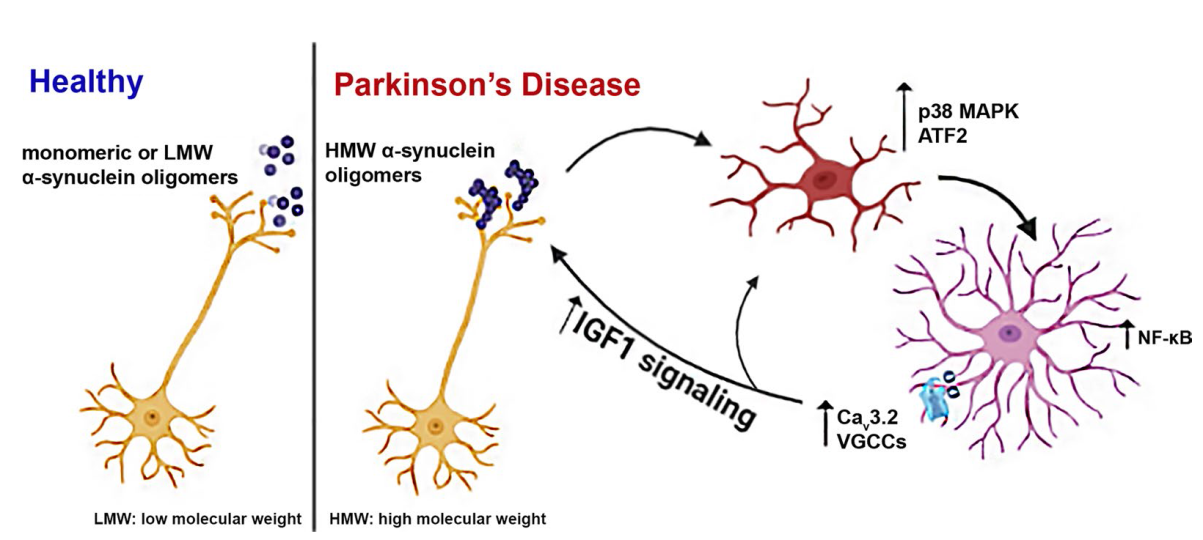
Background It is now realized that Parkinson’s disease (PD) pathology extends beyond the substantia nigra, affecting both central and peripheral nervous systems, and exhibits a variety of non-motor symptoms often preceding motor features. Neuroinflammation induced by activated microglia and astrocytes is thought to underlie these manifestations. α-Synuclein aggregation has been linked with sustained neuroinflammation in PD, aggravating neuronal degeneration; however, there is still a lack of critical information about the structural identity of the α-synuclein conformers that activate microglia and/or astrocytes and the molecular pathways involved.
Methods To investigate the role of α-synuclein conformers in the development and maintenance of neuroinflammation, we used primary quiescent microglia and astrocytes, post-mortem brain tissues from PD patients and A53T α-synuclein transgenic mice that recapitulate key features of PD-related inflammatory responses in the absence of cell death, i.e., increased levels of pro-inflammatory cytokines and complement proteins. Biochemical and -omics techniques including RNAseq and secretomic analyses, combined with 3D reconstruction of individual astrocytes and live calcium imaging, were used to uncover the molecular mechanisms underlying glial responses in the presence of α-synuclein oligomers in vivo and in vitro.
Results We found that the presence of SDS-resistant hyper-phosphorylated α-synuclein oligomers, but not monomers, was correlated with sustained inflammatory responses, such as elevated levels of endogenous antibodies and cytokines and microglial activation. Similar oligomeric α-synuclein species were found in post-mortem human brain samples of PD patients but not control individuals. Detailed analysis revealed a decrease in Iba1Low/CD68Low microglia and robust alterations in astrocyte number and morphology including process retraction. Our data indicated an activation of the p38/ATF2 signaling pathway mostly in microglia and a sustained induction of the NF-κB pathway in astrocytes of A53T mice. The sustained NF-κB activity triggered the upregulation of astrocytic T-type Cav3.2 Ca2+ channels, altering the astrocytic secretome and promoting the secretion of IGFBPL1, an IGF-1 binding protein with anti-inflammatory and neuroprotective potential.
Conclusions Our work supports a causative link between the neuron-produced α-synuclein oligomers and sustained neuroinflammation in vivo and maps the signaling pathways that are stimulated in microglia and astrocytes. It also highlights the recruitment of astrocytic Cav3.2 channels as a potential neuroprotective mediator against the α-synuclein-induced neuroinflammation.
Graphical Abstract
The aetiologies and origins of neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD), are complex and multifaceted. A growing body of evidence suggests that the gut microbiome plays crucial roles in the development and progression of neurodegenerative diseases. Clinicians have come to realize that therapeutics targeting the gut microbiome have the potential to halt the progression of neurodegenerative diseases. This narrative review examines the alterations in the gut microbiome in AD, PD, ALS and HD, highlighting the close relationship between the gut microbiome and the brain in neurodegenerative diseases. Processes that mediate the gut microbiome-brain communication in neurodegenerative diseases, including the immunological, vagus nerve and circulatory pathways, are evaluated. Furthermore, we summarize potential therapeutics for neurodegenerative diseases that modify the gut microbiome and its metabolites, including diets, probiotics and prebiotics, microbial metabolites, antibacterials and faecal microbiome transplantation. Finally, current challenges and future directions are discussed.
Background Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of familial Parkinson’s disease (PD). These mutations elevate the LRRK2 kinase activity, making LRRK2 kinase inhibitors an attractive therapeutic. LRRK2 kinase activity has been consistently linked to specific cell signaling pathways, mostly related to organelle trafficking and homeostasis, but its relationship to PD pathogenesis has been more difficult to define. LRRK2-PD patients consistently present with loss of dopaminergic neurons in the substantia nigra but show variable development of Lewy body or tau tangle pathology. Animal models carrying LRRK2 mutations do not develop robust PD-related phenotypes spontaneously, hampering the assessment of the efficacy of LRRK2 inhibitors against disease processes. We hypothesized that mutations in LRRK2 may not be directly related to a single disease pathway, but instead may elevate the susceptibility to multiple disease processes, depending on the disease trigger. To test this hypothesis, we have previously evaluated progression of α-synuclein and tau pathologies following injection of proteopathic seeds. We demonstrated that transgenic mice overexpressing mutant LRRK2 show alterations in the brain-wide progression of pathology, especially at older ages.
Methods Here, we assess tau pathology progression in relation to long-term LRRK2 kinase inhibition. Wild-type or LRRK2G2019S knock-in mice were injected with tau fibrils and treated with control diet or diet containing LRRK2 kinase inhibitor MLi-2 targeting the IC50 or IC90 of LRRK2 for 3-6 months. Mice were evaluated for tau pathology by brain-wide quantitative pathology in 844 brain regions and subsequent linear diffusion modeling of progression.
Results Consistent with our previous work, we found systemic alterations in the progression of tau pathology in LRRK2G2019S mice, which were most pronounced at 6 months. Importantly, LRRK2 kinase inhibition reversed these effects in LRRK2G2019S mice, but had minimal effect in wild-type mice, suggesting that LRRK2 kinase inhibition is likely to reverse specific disease processes in G2019S mutation carriers. Additional work may be necessary to determine the potential effect in non-carriers.
Conclusions This work supports a protective role of LRRK2 kinase inhibition in G2019S carriers and provides a rational workflow for systematic evaluation of brain-wide phenotypes in therapeutic development.
Proteinopathy, defined as the abnormal accumulation of proteins that eventually leads to cell death, is one of the most significant pathological features of neurodegenerative diseases. Tauopathies, represented by Alzheimer’s disease (AD), and synucleinopathies, represented by Parkinson’s disease (PD), show similarities in multiple aspects. AD manifests extrapyramidal symptoms while dementia is also a major sign of advanced PD. We and other researchers have sequentially shown the cross-seeding phenomenon of α-synuclein (α-syn) and tau, reinforcing pathologies between synucleinopathies and tauopathies. The highly overlapping clinical and pathological features imply shared pathogenic mechanisms between the two groups of disease. The diagnostic and therapeutic strategies seemingly appropriate for one distinct neurodegenerative disease may also apply to a broader spectrum. Therefore, a clear understanding of the overlaps and divergences between tauopathy and synucleinopathy is critical for unraveling the nature of the complicated associations among neurodegenerative diseases. In this review, we discuss the shared and diverse characteristics of tauopathies and synucleinopathies from aspects of genetic causes, clinical manifestations, pathological progression and potential common therapeutic approaches targeting the pathology, in the aim to provide a timely update for setting the scheme of disease classification and provide novel insights into the therapeutic development for neurodegenerative diseases.
Huntington's disease (HD) is a devastating neurodegenerative disorder caused by aggregation of the mutant huntingtin (mHTT) protein, resulting from a CAG repeat expansion in the huntingtin gene HTT. HD is characterized by a variety of debilitating symptoms including involuntary movements, cognitive impairment, and psychiatric disturbances. Despite considerable efforts, effective disease-modifying treatments for HD remain elusive, necessitating exploration of novel therapeutic approaches, including lifestyle modifications that could delay symptom onset and disease progression. Recent studies suggest that time-restricted eating (TRE), a form of intermittent fasting involving daily caloric intake within a limited time window, may hold promise in the treatment of neurodegenerative diseases, including HD. TRE has been shown to improve mitochondrial function, upregulate autophagy, reduce oxidative stress, regulate the sleep-wake cycle, and enhance cognitive function. In this review, we explore the potential therapeutic role of TRE in HD, focusing on its underlying physiological mechanisms. We discuss how TRE might enhance the clearance of mHTT, recover striatal brain-derived neurotrophic factor levels, improve mitochondrial function and stress-response pathways, and synchronize circadian rhythm activity. Understanding these mechanisms is critical for the development of targeted lifestyle interventions to mitigate HD pathology and improve patient outcomes. While the potential benefits of TRE in HD animal models are encouraging, future comprehensive clinical trials will be necessary to evaluate its safety, feasibility, and efficacy in persons with HD.
The renin-angiotensin system (RAS) was classically considered a circulating hormonal system that regulates blood pressure. However, different tissues and organs, including the brain, have a local paracrine RAS. Mutual regulation between the dopaminergic system and RAS has been observed in several tissues. Dysregulation of these interactions leads to renal and cardiovascular diseases, as well as progression of dopaminergic neuron degeneration in a major brain center of dopamine/angiotensin interaction such as the nigrostriatal system. A decrease in the dopaminergic function induces upregulation of the angiotensin type-1 (AT1) receptor activity, leading to recovery of dopamine levels. However, AT1 receptor overactivity in dopaminergic neurons and microglial cells upregulates the cellular NADPH-oxidase-superoxide axis and Ca2+ release, which mediate several key events in oxidative stress, neuroinflammation, and α-synuclein aggregation, involved in Parkinson's disease (PD) pathogenesis. An intraneuronal antioxidative/anti-inflammatory RAS counteracts the effects of the pro-oxidative AT1 receptor overactivity. Consistent with this, an imbalance in RAS activity towards the pro-oxidative/pro-inflammatory AT1 receptor axis has been observed in the substantia nigra and striatum of several animal models of high vulnerability to dopaminergic degeneration. Interestingly, autoantibodies against angiotensin-converting enzyme 2 and AT1 receptors are increased in PD models and PD patients and contribute to blood-brain barrier (BBB) dysregulation and nigrostriatal pro-inflammatory RAS upregulation. Therapeutic strategies addressed to the modulation of brain RAS, by AT1 receptor blockers (ARBs) and/or activation of the antioxidative axis (AT2, Mas receptors), may be neuroprotective for individuals with a high risk of developing PD or in prodromal stages of PD to reduce progression of the disease.
Mitochondria have multiple functions such as supplying energy, regulating the redox status, and producing proteins encoded by an independent genome. They are closely related to the physiology and pathology of many organs and tissues, among which the brain is particularly prominent. The brain demands 20% of the resting metabolic rate and holds highly active mitochondrial activities. Considerable research shows that mitochondria are closely related to brain function, while mitochondrial defects induce or exacerbate pathology in the brain. In this review, we provide comprehensive research advances of mitochondrial biology involved in brain functions, as well as the mitochondria-dependent cellular events in brain physiology and pathology. Furthermore, various perspectives are explored to better identify the mitochondrial roles in neurological diseases and the neurophenotypes of mitochondrial diseases. Finally, mitochondrial therapies are discussed. Mitochondrial-targeting therapeutics are showing great potentials in the treatment of brain diseases.
Background Adult neurogenesis occurs in the subventricular zone (SVZ) and the subgranular zone of the dentate gyrus in the hippocampus. The neuronal stem cells in these two neurogenic niches respond differently to various physiological and pathological stimuli. Recently, we have found that the decrement of carboxypeptidase E (CPE) with aging impairs the maturation of brain-derived neurotrophic factor (BDNF) and neurogenesis in the SVZ. However, it remains unknown whether these events occur in the hippocampus, and what the role of CPE is in the adult hippocampal neurogenesis in the context of Alzheimer’s disease (AD).
Methods In vivo screening was performed to search for miRNA mimics capable of upregulating CPE expression and promoting neurogenesis in both neurogenic niches. Among these, two agomirs were further assessed for their effects on hippocampal neurogenesis in the context of AD. We also explored whether these two agomirs could ameliorate behavioral symptoms and AD pathology in mice, using direct intracerebroventricular injection or by non-invasive intranasal instillation.
Results Restoration of CPE expression in the hippocampus improved BDNF maturation and boosted adult hippocampal neurogenesis. By screening the miRNA mimics targeting the 5’UTR region of Cpe gene, we developed two agomirs that were capable of upregulating CPE expression. The two agomirs significantly rescued adult neurogenesis and cognition, showing multiple beneficial effects against the AD-associated pathologies in APP/PS1 mice. Of note, noninvasive approach via intranasal delivery of these agomirs improved the behavioral and neurocognitive functions of APP/PS1 mice.
Conclusions CPE may regulate adult hippocampal neurogenesis via the CPE-BDNF-TrkB signaling pathway. This study supports the prospect of developing miRNA agomirs targeting CPE as biopharmaceuticals to counteract aging- and disease-related neurological decline in human brains.
The use of biomarker-led clinical trial designs has been transformative for investigating amyloid-targeting therapies for Alzheimer’s disease (AD). The designs have ensured the correct selection of patients on these trials, supported target engagement and have been used to support claims of disease modification and clinical efficacy. Ultimately, this has recently led to approval of disease-modifying, amyloid-targeting therapies for AD; something that should be noted for clinical trials investigating tau-targeting therapies for AD. There is a clear overlap of the purpose of biomarker use at each stage of clinical development between amyloid-targeting and tau-targeting clinical trials. However, there are differences within the potential context of use and interpretation for some biomarkers in particular measurements of amyloid and utility of soluble, phosphorylated tau biomarkers. Given the complexities of tau in health and disease, it is paramount that therapies target disease-relevant tau and, in parallel, appropriate assays of target engagement are developed. Tau positron emission tomography, fluid biomarkers reflecting tau pathology and downstream measures of neurodegeneration will be important both for participant recruitment and for monitoring disease-modification in tau-targeting clinical trials. Bespoke design of biomarker strategies and interpretations for different modalities and tau-based targets should also be considered.
