诊断学理论与实践 ›› 2021, Vol. 20 ›› Issue (03): 245-250.doi: 10.16150/j.1671-2870.2021.03.004
林芙君, 蒋更如( )
)
收稿日期:2021-05-17
出版日期:2021-06-25
发布日期:2022-06-28
通讯作者:
蒋更如
E-mail:jianggeng-ru@hotmail.com
基金资助:
Received:2021-05-17
Online:2021-06-25
Published:2022-06-28
中图分类号:
林芙君, 蒋更如. 奥尔波特综合征的疾病谱扩展对相关疾病诊断、筛查和治疗的启示[J]. 诊断学理论与实践, 2021, 20(03): 245-250.
表1
2018年AS国际工作组提出的AS定义和分型[12]
| 遗传模式 | 突变基因 | 突变类型 | 临床特征 | 进展至ESRD风险 |
|---|---|---|---|---|
| X连锁显性 | COL4A5 | 纯合(男性) | 进展至ESRD的概率以及出现肾外表现的时间受基因型影响 | 100% |
| 杂合(女性) | 影响疾病进展的危险因素包括肉眼血尿、SNHL、蛋白尿、GBM增厚和分层 | 最高可至25% | ||
| 常染色体隐性 | COL4A3或 COL4A4 | 纯合或复合杂合 | 进展至ESRD的概率以及出现肾外表现的时间受基因型影响 | 100% |
| 常染色体显性 | 杂合 | 包括既往被诊断为TBMN/BFH的肾性血尿患者,影响疾病进展的危险因素有蛋白尿、FSGS、GBM增厚和分层、SNHL、患者或家系内成员有疾病进展的证据、合并修饰基因 | 有疾病进展危险因素的患者至少20%,无危险因素的患者<1% | |
| 双基因 | COL4A3、 COL4A4及 COL4A5 | COL4A3和COL4A4 基因反式突变 | 临床表现和遗传模式类似常染色体隐性遗传 | 最高可至100% |
| COL4A3和COL4A4 基因顺式突变 | 临床表现和遗传模式类似常染色体显性遗传 | 最高可至20% | ||
| COL4A5和COL4A3 或COL4A4基因突变 | 不符合孟德尔遗传模式 | 最高可至100% (男性患者) |

图1
AS的疾病谱扩展 XLAS:X连锁显性遗传Alport综合征(X-link Alport syndrome);ARAS:常染色体隐性遗传Alport综合征(autosomal recessive Alport syndrome)。

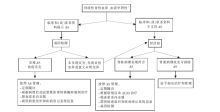
图2
持续性肾性血尿患者的AS筛查流程图[19] *:如血清补体C3、血清补体C4、抗核抗体、抗中性粒细胞胞质抗体、抗肾小球基膜抗体等阴性;血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitor,ACEI)。
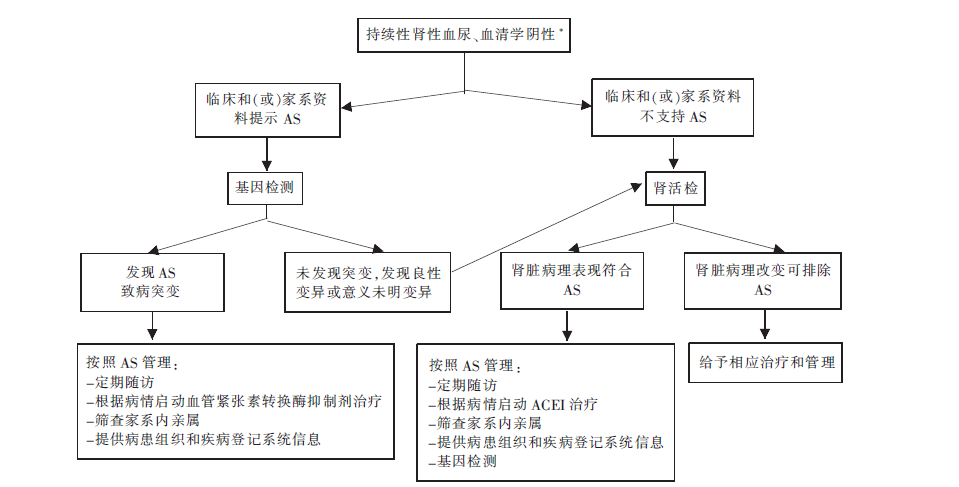
表2
AS的拟表型致病基因和修饰基因变异位点
| 拟表型基因 | 已报道的修饰基因变异位点 |
|---|---|
| MYH9:Fechtner综合征(OMIM155100) COL4A1, COL4A2:HANAC综合征(OMIM611773) CFHR5:电子致密物沉积病(OMIM 134370);CFHR5相关肾病(OMIM 614809) FN1:纤维连接蛋白肾小球病(OMIM601894) LMX1B:指甲-髌骨综合征(OMIM 161200) | MYO1E(c.2627C>G; p.Thr876Arg,c.352A>G; p.Lys118Glu) MYH9(c.4952T>C; p.Met1651Thr) LAMA5(c.3728C>T; p.Pro1243Leu) NPHS2(c.686G>A; p.Arg229Gln) |
| [1] | Alport AC. Hereditary familial congenital haemorrhagic nephritis[J]. Br Med J, 1927, 1(3454):504-506. |
| [2] |
Flinter F. Alport's syndrome[J]. J Med Genet, 1997, 34(4):326-330.
pmid: 9138159 |
| [3] |
Gregory MC, Terreros DA, Barker DF, et al. Alport syndrome--clinical phenotypes, incidence, and pathology[J]. Contrib Nephrol, 1996, 117:1-28.
pmid: 8801040 |
| [4] |
Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy[J]. J Am Soc Nephrol, 2013, 24(3):364-375.
doi: 10.1681/ASN.2012020148 pmid: 23349312 |
| [5] |
ALPORT综合征诊疗共识专家组. Alport综合征诊断和治疗专家推荐意见[J]. 中华肾脏病杂志, 2018, 34(3):227-231.
doi: 10.3760/cma.j.issn.1001-7097.2018.03.014 |
| [6] |
Wang YY, Savige J. The epidemiology of thin basement membrane nephropathy[J]. Semin Nephrol, 2005, 25(3):136-139.
pmid: 15880322 |
| [7] |
Matthaiou A, Poulli T, Deltas C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: a systematic review[J]. Clin Kidney J, 2020, 13(6):1025-1036.
doi: 10.1093/ckj/sfz176 pmid: 33391746 |
| [8] |
Demir E, Caliskan Y. Variations of type Ⅳ collagen-encoding genes in patients with histological diagnosis of focal segmental glomerulosclerosis[J]. Pediatr Nephrol, 2020, 35(6):927-936.
doi: 10.1007/s00467-019-04282-y pmid: 31254113 |
| [9] | Li Y, Groopman EE, D'Agati V, et al. Type Ⅳ collagen mutations in familial IgA nephropathy[J]. Kidney Int Rep, 2020, 5(7):1075-1078. |
| [10] | Barua M, Paterson AD. Population-based studies reveal an additive role of type Ⅳ collagen variants in hematuria and albuminuria[J/OL]. Pediatr Nephrol, 2021-02-26[2021-05-17]. https://pubmed.ncbi.nlm.nih.gov/33635378/. |
| [11] | Furlano M, Martínez V, Pybus M, et al. Clinical and genetic features of autosomal dominant Alport syndrome: a cohort study[J/OL]. Am J Kidney Dis, 2021-04-07[2021-05-17]. https://pubmed.ncbi.nlm.nih.gov/33838161/. |
| [12] |
Kashtan CE, Ding J, Garosi G, et al. Alport syndrome: a unified classification of genetic disorders of collagen Ⅳ α345: a position paper of the Alport Syndrome Classification Working Group[J]. Kidney Int, 2018, 93(5):1045-1051.
doi: 10.1016/j.kint.2017.12.018 URL |
| [13] |
Groopman EE, Marasa M, Cameron-Christie S, et al. Diag-nostic utility of exome sequencing for kidney disease[J]. N Engl J Med, 2019, 380(2):142-151.
doi: 10.1056/NEJMoa1806891 URL |
| [14] | Quinlan C, Rheault MN. Genetic basis of type Ⅳ collagen disorders of the kidney[J/OL]. Clin J Am Soc Nephrol. 2021-04-13[2021-05-17]. https://pubmed.ncbi.nlm.nih.gov/33849932/. |
| [15] |
Gross O, Licht C, Anders HJ, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy[J]. Kidney Int, 2012, 81(5):494-501.
doi: 10.1038/ki.2011.407 URL |
| [16] | Zhang Y, Böckhaus J, Wang F, et al. Genotype-phenotype correlations and nephroprotective effects of RAAS inhibition in patients with autosomal recessive Alport syndrome[J/OL]. Pediatr Nephrol. 2021-03-27[2021-05-17]. https://pubmed.ncbi.nlm.nih.gov/33772369/. |
| [17] |
Gross O, Tönshoff B, Weber LT, et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport′s syndrome[J]. Kidney Int, 2020, 97(6):1275-1286.
doi: 10.1016/j.kint.2019.12.015 URL |
| [18] |
Stock J, Kuenanz J, Glonke N, et al. Prospective study on the potential of RAAS blockade to halt renal disease in Alport syndrome patients with heterozygous mutations[J]. Pediatr Nephrol, 2017, 32(1):131-137.
doi: 10.1007/s00467-016-3452-z URL |
| [19] |
Temme J, Peters F, Lange K, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in hete-rozygous carriers of X-chromosomal and autosomal recessive Alport mutations[J]. Kidney Int, 2012, 81(8):779-783.
doi: 10.1038/ki.2011.452 URL |
| [20] |
Kashtan CE. Alport syndrome: achieving early diagnosis and treatment[J]. Am J Kidney Dis, 2021, 77(2):272-279.
doi: 10.1053/j.ajkd.2020.03.026 URL |
| [21] |
Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020[J]. Pediatr Nephrol, 2021, 36(3):711-719.
doi: 10.1007/s00467-020-04819-6 URL |
| [22] |
Savige J. Should we diagnose autosomal dominant alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant?[J]. Kidney Int Rep, 2018, 3(6):1239-1241.
doi: 10.1016/j.ekir.2018.08.002 pmid: 30450445 |
| [23] | Chan MM, Gale DP. Isolated microscopic haematuria of glomerular origin: clinical significance and diagnosis in the 21st century[J]. Clin Med (Lond), 2015, 15(6):576-580. |
| [24] | Uliana V, Sebastio P, Riva M, et al. Deciphering the pathogenesis of the COL4-related hematuric nephritis: A genotype/phenotype study[J]. Mol Genet Genomic Med, 2021, 9(2):e1576. |
| [25] |
Moreno JA, Yuste C, Gutiérrez E, et al. Haematuria as a risk factor for chronic kidney disease progression in glomerular diseases: a review[J]. Pediatr Nephrol, 2016, 31(4):523-533.
doi: 10.1007/s00467-015-3119-1 URL |
| [26] |
Murray SL, Dorman A, Benson KA, et al. Utility of genomic testing after renal biopsy[J]. Am J Nephrol, 2020, 51(1):43-53.
doi: 10.1159/000504869 pmid: 31822006 |
| [27] | Jayasinghe K, Stark Z, Kerr PG, et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease[J]. Genet Med, 2021, 23(1):183-191. |
| [28] |
Vos P, Zietse R, van Geel M, et al. Diagnosing Alport syndrome: lessons from the pediatric ward[J]. Nephron, 2018, 140(3):203-210.
doi: 10.1159/000492438 URL |
| [29] |
Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases[J]. Nephrol Dial Transplant, 2013, 28(12):2946-2960.
doi: 10.1093/ndt/gft253 URL |
| [30] |
Savige J, Ariani F, Mari F, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome[J]. Pediatr Nephrol, 2019, 34(7):1175-1189.
doi: 10.1007/s00467-018-3985-4 pmid: 29987460 |
| [31] | Savige J, Storey H, Watson E, et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria[J/OL]. Eur J Hum Genet, 2021-04-15[2021-05-17]. https://pubmed.ncbi.nlm.nih.gov/33854215/. |
| [32] | Yamamura T, Nozu K, Minamikawa S, et al. Comparison between conventional and comprehensive sequencing approaches for genetic diagnosis of Alport syndrome[J]. Mol Genet Genomic Med, 2019, 7(9):e883. |
| [33] |
Moreno JA, Sevillano á, Gutiérrez E, et al. Glomerular hematuria: cause or consequence of renal inflammation?[J]. Int J Mol Sci, 2019, 20(9):2205.
doi: 10.3390/ijms20092205 URL |
| [34] |
Bish DR, Bish EK, El-Hajj H, et al. A robust pooled testing approach to expand COVID-19 screening capacity[J]. PLoS One, 2021, 16(2):e0246285.
doi: 10.1371/journal.pone.0246285 URL |
| [35] |
Lin F, Bian F, Zou J, et al. Whole exome sequencing reveals novel COL4A3 and COL4A4 mutations and resolves diagnosis in Chinese families with kidney disease[J]. BMC Nephrol, 2014, 15:175.
doi: 10.1186/1471-2369-15-175 URL |
| [36] |
Zhang Y, Ding J, Wang S, et al. Reassessing the pathogenicity of c.2858G>T(p.(G953V)) in COL4A5 Gene: report of 19 Chinese families[J]. Eur J Hum Genet, 2020, 28(2):244-252.
doi: 10.1038/s41431-019-0523-1 URL |
| [37] |
Shulman C, Liang E, Kamura M, et al. Type Ⅳ collagen variants in CKD: performance of computational predictions for identifying pathogenic variants[J]. Kidney Med, 2021, 3(2):257-266.
doi: 10.1016/j.xkme.2020.12.007 URL |
| [38] |
Omachi K, Kamura M, Teramoto K, et al. A split-luciferase-based trimer formation assay as a high-throughput screening platform for therapeutics in alport syndrome[J]. Cell Chem Biol, 2018, 25(5):634-643.
doi: S2451-9456(18)30043-6 pmid: 29526710 |
| [39] |
Fallerini C, Baldassarri M, Trevisson E, et al. Alport syndrome: impact of digenic inheritance in patients management[J]. Clin Genet, 2017, 92(1):34-44.
doi: 10.1111/cge.12919 pmid: 27859054 |
| [40] |
Horinouchi T, Nozu K, Yamamura T, et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked Alport syndrome[J]. J Am Soc Nephrol, 2018, 29(8):2244-2254.
doi: 10.1681/ASN.2018030228 pmid: 29959198 |
| [41] |
Boeckhaus J, Hoefele J, Riedhammer KM, et al. Precise variant interpretation, phenotype ascertainment, and genotype-phenotype correlation of children in the EARLY PRO-TECT Alport trial[J]. Clin Genet, 2021, 99(1):143-156.
doi: 10.1111/cge.13861 pmid: 33040356 |
| [42] |
Horinouchi T, Yamamura T, Nagano C, et al. Heterozygous urinary abnormality-causing variants of COL4A3 and COL4A4 affect severity of autosomal recessive Alport syndrome[J]. Kidney 360, 2020, 1:936-942.
doi: 10.34067/KID.0000372019 pmid: 35369551 |
| [43] | Yamamura T, Nozu K, Fu XJ, et al. Natural history and genotype-phenotype correlation in female X-linked Alport syndrome[J]. Kidney Int Rep, 2017, 2(5):850-855. |
| [1] | 何亲羽, 王伟, 陈立芬, 张雪蕾, 董治亚. LHCGR基因突变致家族性男性性早熟2例报告及文献复习[J]. 诊断学理论与实践, 2022, 21(05): 598-605. |
| [2] | 陈志敏, 何浩岚. 艾滋病合并马尔尼菲篮状菌病的诊治现状[J]. 诊断学理论与实践, 2022, 21(04): 425-430. |
| [3] | 沈银忠. 《人类免疫缺陷病毒感染/艾滋病合并结核分枝杆菌感染诊治专家共识》解读[J]. 诊断学理论与实践, 2022, 21(04): 431-436. |
| [4] | 施霞, 马鑫, 王珍燕, 张晖, 刘少军. 32例人类免疫缺陷病毒感染合并慢性肾病患者的临床病理特征及随访结果分析[J]. 诊断学理论与实践, 2022, 21(04): 437-443. |
| [5] | 顾炫, 柳俊. 超声筛查鉴别胰腺实性假乳头状瘤与胰腺导管腺癌的研究分析[J]. 诊断学理论与实践, 2022, 21(04): 504-508. |
| [6] | 李娜娜, 齐涛, 朱黎明. 血清胃蛋白酶原、胃泌素17和幽门螺杆菌IgG抗体在胃部疾病初筛中的临床价值[J]. 诊断学理论与实践, 2022, 21(04): 509-513. |
| [7] | 陈宏, 沈银忠. 人类免疫缺陷病毒感染/艾滋病合并结核病的诊治进展[J]. 诊断学理论与实践, 2022, 21(04): 530-534. |
| [8] | 何新, 陈慧, 冯炜炜. 机器学习算法在辅助超声诊断附件肿块良恶性中的应用研究进展[J]. 诊断学理论与实践, 2022, 21(04): 541-546. |
| [9] | 徐子真, 李擎天, 刘湘帆, 李莉, 李惠, 王也飞, 吴洁敏, 陈宁, 梁璆荔, 陈松立, 戴健敏, 宋珍, 丁磊. 实验诊断学在线课程的建立和实践[J]. 诊断学理论与实践, 2022, 21(04): 547-550. |
| [10] | 李佳, 吕良敬. 靶向治疗时代议自身免疫病的感染挑战[J]. 诊断学理论与实践, 2022, 21(03): 299-303. |
| [11] | 赵然, 詹维伟, 侯怡卿. 计算机辅助诊断系统辅助超声诊断甲状腺弥漫性病变合并结节良恶性的应用价值[J]. 诊断学理论与实践, 2022, 21(03): 390-394. |
| [12] | 郭业兵, 郑金峰. 阴道壁胃肠道外间质瘤一例报道并文献复习[J]. 诊断学理论与实践, 2022, 21(03): 405-407. |
| [13] | 中华医学会内分泌学分会. 新型冠状病毒肺炎疫情下骨质疏松症管理专家建议[J]. 诊断学理论与实践, 2022, 21(02): 133-135. |
| [14] | 王刚, 陈生弟. 神经病学的诊断:起源、发展及挑战[J]. 诊断学理论与实践, 2022, 21(01): 1-4. |
| [15] | 唐静仪, 余群, 刘军. 结合人工智能的结构影像分析对阿尔茨海默病的早期预测及精准诊断研究进展[J]. 诊断学理论与实践, 2022, 21(01): 12-17. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||