诊断学理论与实践 ›› 2024, Vol. 23 ›› Issue (06): 568-573.doi: 10.16150/j.1671-2870.2024.06.002
吴健1, 陈玮华1, 徐昊1, 徐元1, 申永秀1, 宁镇2, 秦婷1, 林怡2, 沈鑫2, 郁晓磊2( )
)
收稿日期:2024-06-14
出版日期:2024-12-25
发布日期:2024-12-25
通讯作者:
郁晓磊 E-mail:yuxl1983@163.com基金资助:
WU Jian1, CHEN Weihua1, XU Hao1, XU Yuan1, SHEN Yongxiu1, NING Zhen2, QIN Ting1, LIN Yi2, SHEN Xin2, YU Xiaolei2()
Received:2024-06-14
Published:2024-12-25
Online:2024-12-25
摘要:
目的: 分析上海市人类免疫缺陷病毒(human immunodeficiency virus, HIV)1型(HIV-1)感染者的分子流行特征及影响因素,为制定有效干预措施提供科学依据。方法: 收集2015年至2018年新报告的HIV-1感染者血清样本1 322例,通过巢式PCR扩增HIV-1pol区基因序列,根据pol基因序列进行病毒亚型分析,基于最适基因距离构建分子网络,分析分子网络特征及其影响因素。结果: 1 322例血清样本共获得1 241条HIV-1pol区基因序列,发现12种HIV基因型,以CRF01_AE和CRF07_BC亚型为主,分别占50.93%和34.00%。有530条序列进入分子网络,入网率为42.71%(530/1 241),共形成95个传播簇,簇内节点数为2~219个,其中有一个大簇,节点数为219个(41.32%,219/530)。入网率最高的亚型是CRF07_BC(59.48%,251/422),其次是01B(52.46%,32/61)和CRF01_AE(34.55%,218/631)。男男性行为传播相较于异性性传播更容易形成网络。结论: 2015年至2018年上海市HIV-1基因亚型多样,以CRF01_AE和CRF07_BC亚型为主要优势毒株,分子网络呈聚集性分布,男男性接触者(men who have sex with men,MSM) HIV-1感染者具有更快的病毒传播能力,导致更高的感染率,更易进入网络成为高风险传播者。加强HIV-1分子监测可及时了解毒株在不同人群中的传播动态,可帮助公共卫生部门采取有效的干预措施。
中图分类号:
吴健, 陈玮华, 徐昊, 徐元, 申永秀, 宁镇, 秦婷, 林怡, 沈鑫, 郁晓磊. 2015年至2018年上海市HIV-1感染者基因亚型及分子传播特征分析[J]. 诊断学理论与实践, 2024, 23(06): 568-573.
WU Jian, CHEN Weihua, XU Hao, XU Yuan, SHEN Yongxiu, NING Zhen, QIN Ting, LIN Yi, SHEN Xin, YU Xiaolei. Analysis of gene subtypes and molecular transmission characteristics of HIV-1 infected individuals in Shanghai from 2015 to 2018[J]. Journal of Diagnostics Concepts & Practice, 2024, 23(06): 568-573.
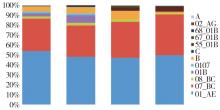
图1
上海市2015-2018年HIV-1感染者亚型分布
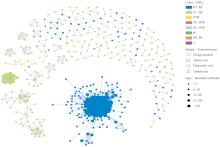
图2
基于FastTree和HIV-Trace构建的分子网络图

表1
2015-2018年上海市HIV-1感染者分子网络入网率影响因素分析
| Variables | Sample size | 2015 | 2016 | 2017 | 2018 | Clustered | Non-clustered | χ2 | P value | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Count | Proportion | Count | Proportion | |||||||||
| Age at diagnosis | 1.348 | 0.72 | ||||||||||
| ≤24 | 234 | 103 | 56 | 37 | 38 | 101 | 43.16% | 133 | 56.84% | |||
| 25-39 | 647 | 299 | 159 | 94 | 95 | 283 | 43.74% | 364 | 56.26% | |||
| 40-54 | 230 | 103 | 67 | 38 | 22 | 96 | 41.74% | 134 | 58.26% | |||
| ≥55 | 130 | 55 | 39 | 26 | 10 | 50 | 38.46% | 80 | 61.54% | |||
| Sex | 8.82 | 0.003 | ||||||||||
| Male | 1168 | 526 | 295 | 182 | 165 | 511 | 43.75% | 657 | 56.25% | |||
| Female | 73 | 34 | 26 | 13 | 0 | 19 | 26.03% | 54 | 73.97% | |||
| Registered residence | 0.6138 | 0.43 | ||||||||||
| Shanghai | 432 | 201 | 121 | 69 | 41 | 191 | 44.21% | 241 | 55.79% | |||
| Other provinces | 809 | 359 | 200 | 126 | 124 | 339 | 41.90% | 470 | 58.10% | |||
| Ethnicity | 7.466 | 0.006 | ||||||||||
| Han | 1200 | 533 | 315 | 192 | 160 | 521 | 43.42% | 679 | 56.58% | |||
| Others | 41 | 27 | 6 | 3 | 5 | 9 | 21.95% | 32 | 78.05% | |||
| Marital status | 2.586 | 0.46 | ||||||||||
| Unmarried | 835 | 369 | 209 | 125 | 132 | 362 | 43.35% | 473 | 56.65% | |||
| Married | 266 | 132 | 67 | 44 | 23 | 110 | 41.35% | 156 | 58.65% | |||
| Divorced or widowed | 137 | 59 | 45 | 24 | 9 | 58 | 42.34% | 79 | 57.66% | |||
| Unknown | 3 | 0 | 0 | 2 | 1 | 0 | 0.00% | 3 | 100.00% | |||
| Education level | 4.773 | 0.44 | ||||||||||
| Illiterate | 11 | 6 | 2 | 3 | 0 | 3 | 27.27% | 8 | 72.73% | |||
| Primary school | 79 | 50 | 18 | 7 | 4 | 27 | 34.18% | 52 | 65.82% | |||
| Junior high school | 253 | 125 | 78 | 38 | 12 | 110 | 43.48% | 143 | 56.52% | |||
| High School | 299 | 140 | 73 | 47 | 39 | 126 | 42.14% | 173 | 57.86% | |||
| College and above | 600 | 239 | 150 | 100 | 110 | 264 | 44.07% | 335 | 55.93% | |||
| Transmission route | 19.55 | <0.001 | ||||||||||
| Transmission route | 884 | 372 | 231 | 135 | 146 | 407 | 46.04% | 477 | 53.96% | |||
| Transmission route | 309 | 158 | 85 | 52 | 14 | 114 | 36.89% | 195 | 63.11% | |||
| Heterosexual | 43 | 30 | 5 | 4 | 4 | 8 | 18.60% | 35 | 81.40% | |||
| Injection drug use | 5 | 0 | 0 | 4 | 1 | 1 | 20.00% | 4 | 80.00% | |||
| Subtype | 74.57 | <0.001 | ||||||||||
| CRF01_AE | 632 | 303 | 155 | 92 | 82 | 218 | 34.65% | 413 | 65.35% | |||
| CRF 07_BC | 422 | 182 | 101 | 71 | 68 | 251 | 59.48% | 171 | 40.52% | |||
| CRF 08_BC | 28 | 13 | 8 | 5 | 2 | 7 | 25.00% | 21 | 75.00% | |||
| Others | 159 | 62 | 57 | 27 | 13 | 53 | 33.33% | 106 | 66.67% | |||
| [1] |
SMITH D M, MAY S J, TWEETEN S, et al. A public health model for the molecular surveillance of HIV transmission in San Diego, California[J]. AIDS, 2009, 23(2):225-232.
doi: 10.1097/QAD.0b013e32831d2a81 pmid: 19098493 |
| [2] | LITTLE S J, KOSAKOVSKY POND S L, ANDERSON C M, et al. Using HIV networks to inform real time prevention interventions[J]. PLoS One, 2014, 9(6):e98443. |
| [3] | 甘梦泽, 冯毅, 邢辉. 基于分子网络方法研究HIV感染者传播特征的相关进展[J]. 中华流行病学杂志, 2019, 40(11):1487-1491. |
| GAN M Z, FENG Y, XING H. Progress in research on the transmission characteristics of HIV-infected persons based on molecular network method[J]. Chin J Epidemiol, 2019, 40(11):1487-1491. | |
| [4] | 冯毅, 王栋, 邢辉. HIV分子传播网络助力我国艾滋病精准防控[J]. 中国艾滋病性病, 2023, 29(1):1-8. |
| FENG Y, WANG D, XING H. HIV molecular transmission network helps precise prevention and control of AIDS in China[J]. Chin J AIDS & STD, 2023, 29(1):1-8. | |
| [5] | 李一, 惠珊, 张锦慧, 等. 黑龙江省2018年新发现HIV-1感染者毒株亚型分子簇及耐药情况[J]. 中国艾滋病性病, 2021, 27(7):680-684. |
| LI Y, HUI S, ZHANG J H, et al. Analysis on HIV subtypes,molecular clusters and drug resistance among newly reported infections in Heilongjiang Province in 2018[J]. Chin J AIDS & STD, 2021, 27(7):680-684. | |
| [6] | NATIONAL CENTER FOR AIDS/STD CONTROL AND PREVENTION C C F D C A P. Guideline of monitor and intervention technique of HIV transmission network in China has been issued[Z]. 2021. |
| [7] | HAO J, ZHENG S, GAN M, et al. Changing proportions of HIV-1 subtypes and transmitted drug resistance among newly diagnosed HIV/AIDS individuals - China, 2015 and 2018[J]. China CDC Wkly, 2021, 3(53):1133-1138. |
| [8] | FAN Q, ZHANG J, LUO M, et al. Molecular genetics and epidemiological characteristics of HIV-1 epidemic strains in various sexual risk behaviour groups in developed Eastern China, 2017-2020[J]. Emerg Microbes Infect, 2022, 11(1):2326-2339. |
| [9] | 郭琪, 王慧, 齐晓晨, 等. 吉林省HIV-1感染者传播簇和分子网络特征研究[J]. 中国卫生工程学, 2023, 22(06):732-736. |
| GUO Q, WANG H, QI X C, et al. Molecular transmission cluster and network characteristics of untreated HIV-1 infected persons in Jilin Orovince[J]. Chin J Public Health Eng, 2023, 22(06):732-736. | |
| [10] | 宋玲, 杨东智, 曹慜, 等. 2012-2020年宁夏HIV-1感染者分子传播网络特征分析[J]. 宁夏医学杂志, 2022, 44(6):504-508. |
| SONG L, YANG D Z, CAO M, et al. Analysis of molecular transmission network characteristics of HIV-1 infected person in Ningxia from 2012 to 2020[J]. Ningxia Med J, 2022, 44(6):504-508. | |
| [11] | 江河, 唐凯玲, 黄精华, 等. 基于分子网络的广西壮族自治区HIV传播热点和跨地区传播特征分析[J]. 中华流行病学杂志, 2022, 43(9):1423-1429. |
| JIANG H, TANG K L, HUANG J H, et al. Analysis of HIV transmission hotspots and characteristics of cross-regional transmission in Guangxi Zhuang Autonomous Region based on molecular network[J]. Chin J Epidemiol, 2022, 43(9):1423-1429. | |
| [12] | 朱可心, 庄勋, 周元, 等. 人类免疫缺陷病毒-1 CRF01 AE 毒株在我国的分子流行特征分析[J]. 中华传染病杂志, 2015(7):396-402. |
| ZHU K X, ZHUANG X, ZHOU Y, et al. Molecular epidemiological characterization of human immunodeficiency virus-1 CRF01 AE strains in China[J]. Chin J Infect Dis, 2015(7):396-402. | |
| [13] | 林怡, 薛以乐, 王绪琴, 等. 上海市男男性行为者中感染HIV-1的CRF01_AE毒株传播簇分析[J]. 中国艾滋病性病, 2016, 22(3):165-169. |
| LIN Y, XUE Y L, WANG X Q, et al. HIV-1 transmission clusters among men who have sex with men in Shanghai[J]. Chin J AIDS STD, 2016, 22(3):165-169. | |
| [14] | 王栋, 冯毅, 郝静静, 等. 基于HIV-1的pol基因区序列分析我国CRF07_BC毒株的跨地区传播特征[J]. 中国艾滋病性病, 2024, 30(3):232-239. |
| WANG D, FENG Y, HAO J J, et al. Characterization of cross-regional transmission on the predominantly endemic CRF07_BC strain in China through the sequence of the pol gene region of HIV-1[J]. Chin J AIDS STD, 2024, 30(3):232-239. | |
| [15] | GAN M, ZHENG S, HAO J, et al. Spatiotemporal patterns of CRF07_BC in China: A population-based study of the HIV strain with the highest infection rates[J]. Front Immunol, 2022, 13:824178. |
| [16] | 刘欢, 金怡晨, 汤后林. HIV感染者的高危行为接触者追踪研究进展[J]. 中华流行病学杂志, 2024, 45(5):755-760. |
| LIU H, JIN Y C, TAN H L. Progress in research of tra-cing contacts exposed to high risk behaviors of HIV-infected patients[J]. Chin J Epidemiol, 2024, 45(5):755-760. | |
| [17] | 赖静兰, 陈雅红, 叶寒辉, 等. 艾滋病合并马尔尼菲篮状菌病患者贫血特征分析[J]. 中国临床研究, 2023, 36(11):1691-1694. |
| LAI JL, CHEN YH, YE H H, et al. Analysis of anemia characteristics of AIDS patients with Marneffei's basket shaped mycosis[J]. Chin Clin Res, 2023, 36(11):1691-1694. | |
| [18] | 宋明明, 张晓, 刘冬莹, 等. HIV/AIDS患者更换二线抗病毒治疗药物效果评价[J]. 中国临床研究, 2023, 36(10):1509-1512. |
| SONG M M, ZHANG X, LIU D Y, et al. Evaluation of the effectiveness of switching second-line antiviral drugs in HIV/AIDS patients[J]. Chin Clin Res, 2023, 36(10):1509-1512. |
| [1] | 胡静静, 沈银忠, 刘莉, 卢洪洲. 艾滋病合并播散性非结核分枝杆菌病诊治现状及研究进展[J]. 诊断学理论与实践, 2023, 22(04): 402-406. |
| [2] | 陈宏, 沈银忠. 人类免疫缺陷病毒感染/艾滋病合并结核病的诊治进展[J]. 诊断学理论与实践, 2022, 21(04): 530-534. |
| [3] | 沈银忠. 《人类免疫缺陷病毒感染/艾滋病合并结核分枝杆菌感染诊治专家共识》解读[J]. 诊断学理论与实践, 2022, 21(04): 431-436. |
| [4] | 郭静, 沈银忠. HIV感染/AIDS合并结核病的临床及免疫学特点研究进展[J]. 诊断学理论与实践, 2021, 20(04): 401-406. |
| [5] | 张林, 周莹, 李海聪, 孙美艳, 孙文秀, 陆玲庆, 朱召芹, 唐琪, 张仁芳, 沈银忠, 卢洪洲. HIV病毒过滤器过滤效率的评估实验研究[J]. 诊断学理论与实践, 2019, 18(06): 676-679. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||