诊断学理论与实践 ›› 2022, Vol. 21 ›› Issue (05): 592-597.doi: 10.16150/j.1671-2870.2022.05.008
宋宗先, 董治亚, 陆子文, 李彦晓, 陈烨( )
)
收稿日期:2022-01-19
出版日期:2022-10-25
发布日期:2023-01-29
通讯作者:
陈烨
E-mail:chenye054@126.com
SONG Zongxian, DONG Zhiya, LU Ziwen, LI Yanxiao, CHEN Ye()
Received:2022-01-19
Online:2022-10-25
Published:2023-01-29
Contact:
CHEN Ye
E-mail:chenye054@126.com
摘要:
目的:分析先天性全身脂肪营养不良1型(congenital generalized lipodystrophy type 1, CGL1)患者的基因型和临床表型,提高临床对该疾病的认识。方法:分析1例因严重黑棘皮就诊的CGL1患儿的临床资料、实验室检查及基因检测结果,并结合现有文献报道的5例中国CGL1病例进行分析、总结。结果:患儿为10岁11个月的女性,临床表现为严重的黑皮棘伴有皮下脂肪减少。实验室检查显示高胰岛素血症、糖耐量异常、骨龄增大、脂联素降低、多囊卵巢。基因检测提示患儿存在AGPAT2基因复合杂合变异,c.646A>T:p.K216*(PVS1_Strong+ PM2+ PM3,来自父亲),c.406G>A:p.G136R(PM3_Strong+ PM1+ PM2+ PP3,来自母亲),2个位点均为可能致病的突变位点,在中国知网数据库中均未见该位点报道。文献复习显示,我国目前报道5例CGL1病例(其中男性2例,女性3例,年龄在66 d到26岁之间),所有患者均存在皮下脂肪减少,有2例患者存在较为严重的黑棘皮症状。结论:CGL1在我国人群中极为罕见,黑棘皮伴皮下脂肪减少需考虑此病,该病的精准诊断有赖于基因检测。
中图分类号:
宋宗先, 董治亚, 陆子文, 李彦晓, 陈烨. 先天性全身脂肪营养不良1型患者的基因型和临床表型分析及文献复习[J]. 诊断学理论与实践, 2022, 21(05): 592-597.
SONG Zongxian, DONG Zhiya, LU Ziwen, LI Yanxiao, CHEN Ye. Genotype and clinical phenotype in congenital generalized lipodystrophy type 1: analysis and literature review[J]. Journal of Diagnostics Concepts & Practice, 2022, 21(05): 592-597.
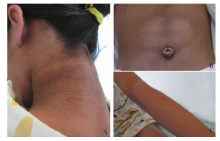
图1
患儿体征 A:颈部黑棘皮;B:腹肌轮廓突出;C:多毛
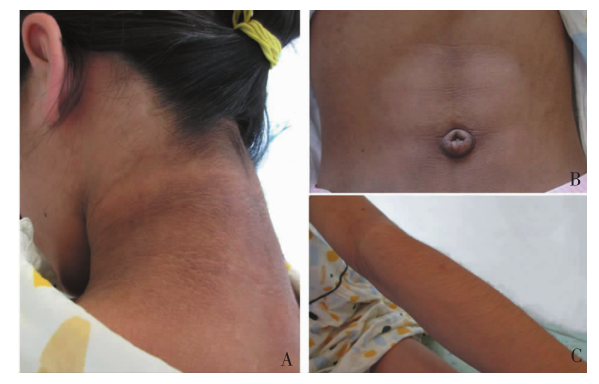
表1
患儿口服葡萄糖耐量试验及同步血浆胰岛素、C肽释放试验
| 时间(min) | 血糖(mmol/L) | 胰岛素(μIU/mL) | C肽(μg/L) |
|---|---|---|---|
| 0 | 4.49 | 66.03↑ | 5.07↑ |
| 30 | 6.70 | 379.00 | 14.60 |
| 60 | 8.07 | 577.70 | 19.44 |
| 120 | 9.08 | >1 000 | 30.51 |
| 180 | 2.30 | 747.50 | 19.99 |

图2
患儿及其父母AGPAT2基因Sanger测序结果 A:红色箭头指示患儿及其父亲c.646位点的腺嘌呤(A)被胸腺嘧啶(T)取代,产生无义突变,从而导致提前出现终止密码子;B:患儿及其母亲从c.406位点的鸟嘌呤(G)被腺嘌呤(A)取代,导致错义突变,谷氨酸被精氨酸替代。
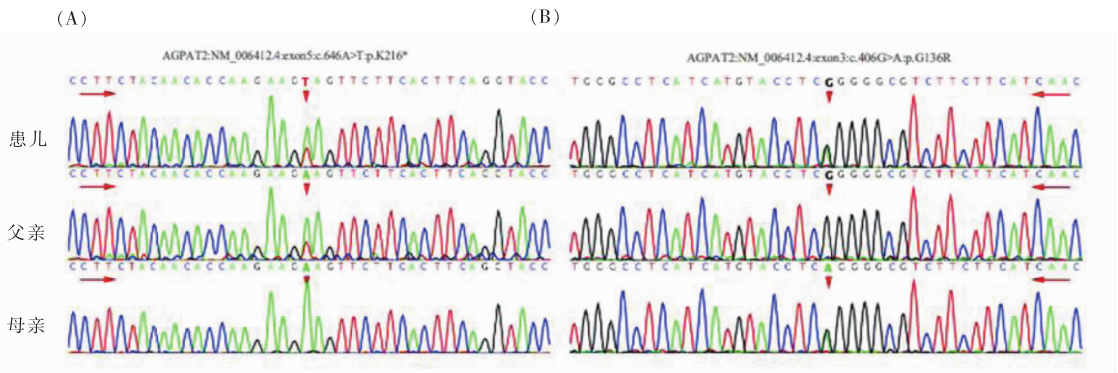
表2
AGPAT2基因突变的临床特点及基因表型
| 病例来源 | Liu等[ | 郭丹丹等[ | 王怡萍等[ | 孟源源等[ | 张娟等[ | 本例 |
|---|---|---|---|---|---|---|
| 报道年份 | 2019 | 2020 | 2020 | 2020 | 2021 | 2021 |
| 性别 | 男 | 男 | 女 | 女 | 女 | 女 |
| 年龄 | 16个月 | 66 d | 4个月 | 12岁 | 26岁 | 10岁11个月 |
| BMI(kg/m2) | 18.3 | 15.4 | 15.8 | 17.1 | 21.8 | 19.8 |
| 主诉 | 发育迟缓 | 皮下结节、腹胀 | 体重增长慢 | 多尿,消瘦 | 糖尿病9年 | 黑棘皮 |
| 特殊面容 | 无 | 无 | 无 | 三角脸 | 无 | 无 |
| 脂肪萎缩肌肉明显 | 是 | 是 | 是 | 是 | 是 | 是 |
| 肝肿大 | 是 | 是 | 是 | 是 | 是 | 否 |
| 糖尿病 | 否 | 是 | 否 | 是 | 是 | 否 |
| 高甘油三酯症 | 是 | 是 | 否 | 是 | 是 | 否 |
| 胰岛素抵抗症状 | 无 | 无 | 无 | 黑棘皮,多毛 | 黑棘皮,多毛 | 黑棘皮,多毛 |
| 其他 | 无 | 皮下结节 | 无 | 右肾积水甲状腺 多发胶质结节 | 卵巢囊肿肝血管瘤 | 骨龄大多囊卵巢 |
| 空腹血糖(mmol/L) | 3.9 | 16.99 | 正常 | 13.5 | 升高 | 4.49 |
| 空腹胰岛素(μIU/mL) | - | - | 正常 | - | 升高 | 66.03 |
| 空腹C肽(μg/L) | - | 20.75 | - | 2.15 | 升高 | 5.07 |
| 糖化血红蛋白(%) | - | 6.3 | - | 14.7 | 8.6 | 5.5 |
| 瘦素(ng/mL) | - | - | - | - | - | 2.17 |
| 脂联素(g/mL) | - | - | - | - | - | 0.28 |
| AGPAT2突变位点 | c.513delC / c.622_626del TCCTC | c.646A>T 纯合突变 | c.335C>T/ c.792_805del GGAGAACGGGGCCA | c.379G>C/ c.317-10T>A | c.356_362del/ c.683T>C | c.646A>T/c.406G>A |
| 氨基酸 | Glu172Argfs*81/ Ser208Leufs*267 | Lys216Ter | Pro112Leu/ Gln264Hisfs*208 | Gly127Arg/splicing | Ala119fs/Leu228Pro | Lys216Ter/ Gly136Arg |
| [1] |
Agarwal AK, Simha V, Oral EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy[J]. J Clin Endocrinol Metab, 2003, 88(10):4840-4847.
doi: 10.1210/jc.2003-030855 URL |
| [2] |
Akinci B, Onay H, Demir T, et al. Natural History of Congenital Generalized Lipodystrophy: A Nationwide Study From Turkey[J]. J Clin Endocrinol Metab, 2016, 101(7):2759-2767.
doi: 10.1210/jc.2016-1005 pmid: 27144933 |
| [3] |
Magré J, Delépine M, Van Maldergem L, et al. Prevalence of mutations in AGPAT2 among human lipodystrophies[J]. Diabetes, 2003, 52(6):1573-1578.
doi: 10.2337/diabetes.52.6.1573 pmid: 12765973 |
| [4] | 郭丹丹, 刘小凤, 段元冬. 66日龄婴儿发现多发皮下结节1月余[J]. 中国当代儿科杂志, 2020, 22(8):903-908. |
| Guo DD, Liu XF, Duan YD. Multiple subcutaneous no-dules for 46 days in an infant aged 66 days[J]. Chin J Contemp Pediatr, 2020, 22(8):903-908. | |
| [5] |
Agarwal AK, Arioglu E, de Almeida S, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34[J]. Nat Genet, 2002, 31(1):21-23.
doi: 10.1038/ng880 pmid: 11967537 |
| [6] |
Liu Y, Li D, Ding Y, et al. Further delineation of AGPAT2 and BSCL2 related congenital generalized lipodystrophy in young infants[J]. Eur J Med Genet, 2019, 62(9):103542.
doi: 10.1016/j.ejmg.2018.09.009 URL |
| [7] | 王怡萍, 朱彦丽, 白晋丽, 等. AGPAT2基因新变异导致先天性全身性脂肪代谢障碍Ⅰ型一例[J]. 中华医学遗传学杂志, 2020, 37(10):1158-1161. |
| Wang YP, Zhu YL, Bai JL, et al. Identification of a novel AGPAT2 variant in a Chinese patient with congenital generalized lipodystrophy type 1[J]. Chin J Med Genet, 2020, 37(10):1158-1161. | |
| [8] | 孟源源, 吴蔚, 黄轲, 等. AGPAT2基因变异致先天性全身脂肪营养不良症1例报告并文献复习[J]. 临床儿科杂志, 2020, 38(8):599-602. |
| Meng YY, Wu W, Huang K, et al. Congenital generalized lipodystrophy caused by mutation of AGPAT2 gene: a case report and literature review[J]. J Chin Pediatr, 2020, 38(8):599-602. | |
| [9] | 张娟, 章钟允, 李浩榕, 等. AGPAT2基因复合杂合突变导致全身性脂肪营养不良1型一例并文献复习[J]. 中华内分泌代谢杂志, 2021, 37(9):840-844. |
| Zhang J, Zhang ZY, Li HR, et al. Generalized lipodystrophy type 1 due to compound heterozygous mutation of AGPAT2 gene: One case report and literature teview[J]. Chin J Endocrinol Metab, 2021, 37(9):840-844. | |
| [10] |
Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction[J]. Nat Rev Endocrinol, 2015, 11(9):522-534.
doi: 10.1038/nrendo.2015.123 pmid: 26239609 |
| [11] |
Lima JG, Nobrega LH, de Lima NN, et al. linical and laboratory data of a large series of patients with congenital generalized lipodystrophy[J]. Diabetol Metab Syndr, 2016, 8:23.
doi: 10.1186/s13098-016-0140-x URL |
| [12] |
Garg A. Acquired and inherited lipodystrophies[J]. N Engl J Med, 2004, 350(12):1220-1234.
doi: 10.1056/NEJMra025261 URL |
| [13] |
Garg A. Lipodystrophies[J]. Am J Med, 2000, 108(2):143-152.
doi: 10.1016/s0002-9343(99)00414-3 pmid: 11126308 |
| [14] | Ren M, Shi J, Jia J, et al. Genotype-phenotype correlations of Berardinelli-Seip congenital lipodystrophy and novel candidate genes prediction[J]. OOrphanet J Rare Dis, 2020, 15(1):108. |
| [15] |
Subauste AR, Das AK, Li X, et al. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations[J]. Diabetes, 2012, 61(11):2922-2931.
doi: 10.2337/db12-0004 pmid: 22872237 |
| [16] |
Vettor R, Milan G, Rossato M, et al. Review article: adipocytokines and insulin resistance[J]. Aliment Pharmacol Ther, 2005, 22(Suppl 2):3-10.
doi: 10.1111/j.1365-2036.2005.02587.x URL |
| [17] |
Antuna-Puente B, Boutet E, Vigouroux C, et al. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-o-acyltransferase-2 deficiency[J]. J Clin Endocrinol Metab, 2010, 95(3):1463-1468.
doi: 10.1210/jc.2009-1824 pmid: 20097706 |
| [18] |
Musso C, Cochran E, Javor E, et al. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients[J]. Metabolism, 2005, 54(2):255-263.
doi: 10.1016/j.metabol.2004.08.021 URL |
| [19] |
Araújo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: a step-by-step approach[J]. J Endocrinol Invest, 2019, 42(1):61-73.
doi: 10.1007/s40618-018-0887-z pmid: 29704234 |
| [1] | 张娟娟, 张婉玉, 韩晓伟, 肖园, 陆文丽, 吕圣. FGFR3基因R248C突变致不成比例矮小伴特殊面容和黑棘皮病一例及文献复习[J]. 诊断学理论与实践, 2020, 19(02): 129-134. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||