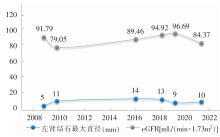
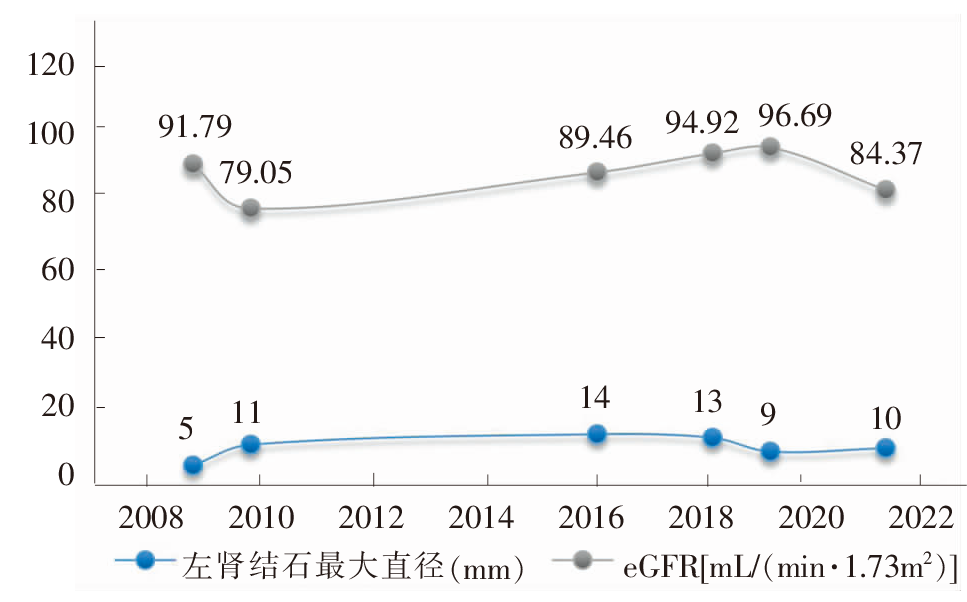
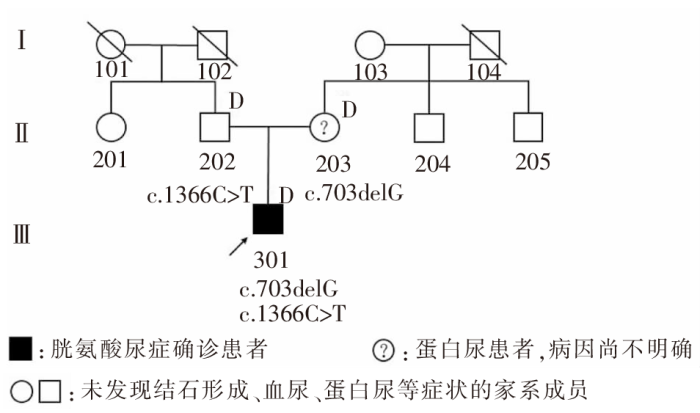
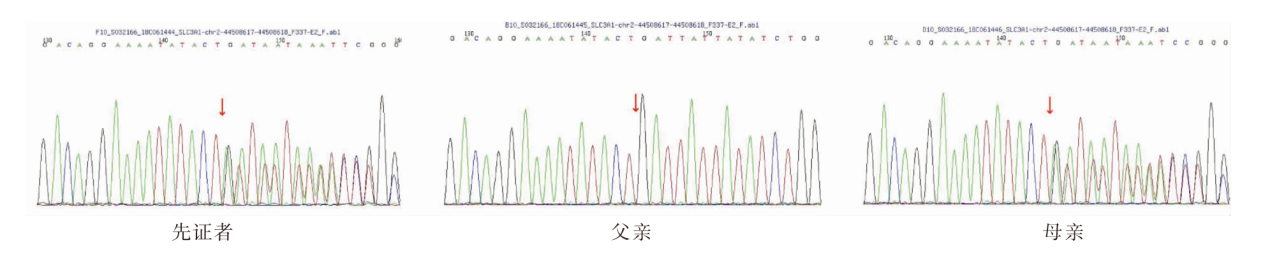
| [1] |
Eisner BH, Goldfarb DS, Baum MA, et al. Evaluation and medical management of patients with cystine nephrolithiasis[J]. J Endourol, 2020, 34(11): 1103-1110.
doi: 10.1089/end.2019.0703
URL
|
| [2] |
Li C, Yang Y, Zheng Y, et al. Genetic and clinical analyses of 13 Chinese families with cystine urolithiasis and identification of 15 novel pathogenic variants in SLC3A1 and SLC7A9[J]. Front Genet, 2020, 11: 74.
doi: 10.3389/fgene.2020.00074
URL
|
| [3] |
Rhodes HL, Yarram-Smith L, Rice SJ, et al. Clinical and genetic analysis of patients with cystinuria in the United Kingdom[J]. Clin J Am Soc Nephrol, 2015, 10(7): 1235-1245.
doi: 10.2215/CJN.10981114
URL
|
| [4] |
Servais A, Thomas K, Dello Strologo L, et al. Cystinuria: clinical practice recommendation[J]. Kidney Int, 2021, 99(1): 48-58.
doi: 10.1016/j.kint.2020.06.035
pmid: 32918941
|
| [5] |
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
doi: 10.1038/gim.2015.30
pmid: 25741868
|
| [6] |
Sahota A, Tischfield JA, Goldfarb DS, et al. Cystinuria: genetic aspects, mouse models, and a new approach to therapy[J]. Urolithiasis, 2019, 47(1): 57-66.
doi: 10.1007/s00240-018-1101-7
pmid: 30515543
|
| [7] |
Font-Llitjós M, Jiménez-Vidal M, Bisceglia L, et al. New insights into cystinuria: 40 new mutations, genotype-phenotype correlation, and digenic inheritance causing partial phenotype[J]. J Med Genet, 2005, 42(1): 58-68.
pmid: 15635077
|
| [8] |
Alghamdi M, Alhasan KA, Taha Elawad A, et al. Diversity of phenotype and genetic etiology of 23 cystinuria saudi patients[J]. Front Pediatr, 2020, 8: 569389.
doi: 10.3389/fped.2020.569389
URL
|
| [9] |
Daga A, Majmundar AJ, Braun DA, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis[J]. Kidney Int, 2018, 93(1): 204-213.
doi: 10.1016/j.kint.2017.06.025
URL
|
| [10] |
Daga S, Palit V, Forster JA, et al. An update on evaluation and management in cystinuria[J]. Urology, 2021, 149: 70-75.
doi: 10.1016/j.urology.2020.12.025
pmid: 33421442
|
| [11] |
Rajiah P, Parakh A, Kay F, et al. Update on multienergy CT: physics, principles, and applications[J]. 2020, 40(5): 1284-1308.
|
| [12] |
张玉娇, 李小虎, 江兆翔, 等. 双能量能谱CT分析体内尿酸结石的诊断价值[J]. 安徽医科大学学报, 2017, 52(7): 1901-1904.
|
| [13] |
Rompsaithong U, Jongjitaree K, Korpraphong P, et al. Characterization of renal stone composition by using fast kilovoltage switching dual-energy computed tomography compared to laboratory stone analysis[J]. Abdom Radiol (NY), 2019, 44(3): 1027-1032.
doi: 10.1007/s00261-018-1787-6
pmid: 30259102
|
| [14] |
Shah TT, Gao C, Peters M, et al. Factors associated with spontaneous stone passage in a contemporary cohort of patients presenting with acute ureteric colic[J]. BJU Int, 2019, 124(3): 504-513.
doi: 10.1111/bju.14777
URL
|
| [15] |
Thomas K, Wong K, Withington J, et al. Cystinuria—a urologist’s perspective[J]. Nat Rev Urol, 2014, 11(5):270-277.
doi: 10.1038/nrurol.2014.51
pmid: 24662732
|
| [16] |
Chillarón J, Font-Llitjós M, Fort J, et al. Pathophysiology and treatment of cystinuria[J]. Nat Rev Nephrol, 2010, 6(7): 424-434.
doi: 10.1038/nrneph.2010.69
pmid: 20517292
|
 ), 王朝晖1a(
), 王朝晖1a(