诊断学理论与实践 ›› 2019, Vol. 18 ›› Issue (03): 263-270.doi: 10.16150/j.1671-2870.2019.03.005
汪婷婷, 郑乃盛, 袁向亮( ), 沈立松()
), 沈立松()
收稿日期:2019-02-21
出版日期:2019-06-25
发布日期:2019-06-25
通讯作者:
袁向亮,沈立松
E-mail:yuanxiangliang@gmail.com;lisongshen@hotmail.com
WANG Tingting, ZHENG Naisheng, YUAN Xiangliang(), SHEN Lisong()
Received:2019-02-21
Online:2019-06-25
Published:2019-06-25
Contact:
YUAN Xiangliang,SHEN Lisong
E-mail:yuanxiangliang@gmail.com;lisongshen@hotmail.com
摘要:
目的: 明确溃疡性结肠炎(ulcerative colitis,UC)与肠道菌群间的相互关系,为寻找简单、安全的UC治疗方法提供实验基础。方法: 通过3%硫酸葡聚糖钠(dextran sulfate sodium,DSS)诱导建立小鼠溃疡性结肠炎模型,观察检测体重变化、肠道病理改变,采用16S rRNA高通量测序技术,检测小鼠粪便中的肠道菌群,并分析比较结肠炎小鼠与正常小鼠间肠道菌群的多样性及丰度。同时,利用已有数据库,预估肠道菌群内的功能基因构成,分析2组间的菌群基因功能差异。结果: 溃疡性结肠炎小鼠体重明显减轻,且肠道上皮完整性被破坏,同时肠道菌群多样性明显降低,溃疡性肠炎组小鼠肠道内双歧杆菌属降至0.2%明显低于正常组小鼠(P<0.05),而乳酸杆菌属平均丰度也显著降低至2.9%(P<0.05),提示可能是通过影响机体代谢从而造成溃疡性结肠炎的发生、发展。结论: 溃疡性结肠炎小鼠的肠道菌群多样性降低,菌群分布均发生显著改变。
中图分类号:
汪婷婷, 郑乃盛, 袁向亮, 沈立松. 基于16S rRNA高通量测序技术分析小鼠实验性结肠炎肠道菌群结构特征[J]. 诊断学理论与实践, 2019, 18(03): 263-270.
WANG Tingting, ZHENG Naisheng, YUAN Xiangliang, SHEN Lisong. Analysis of structural characteristics of gut microbiome in colitis mice based on 16S rRNA high-throughput sequencing[J]. Journal of Diagnostics Concepts & Practice, 2019, 18(03): 263-270.
表1
结肠黏膜损伤指数评分标准
| 指标 | 评分(分) |
|---|---|
| 炎症程度 无 轻度 重度 | 0 1 2 |
| 炎症累及范围 1%~25% 26%~50% 51%~75% 75%~100% | 1 2 3 4 |
| 增生程度 无 轻度 重度 | 0 1 2 |
| 病变深度 无 黏膜下层 肌层 浆膜层 | 0 1 2 3 |
| 结肠溃疡 无 小(≤3 mm) 大(>3 mm) | 0 1 2 |
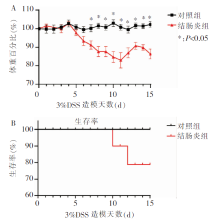
图1
2组小鼠体重及生存率比较
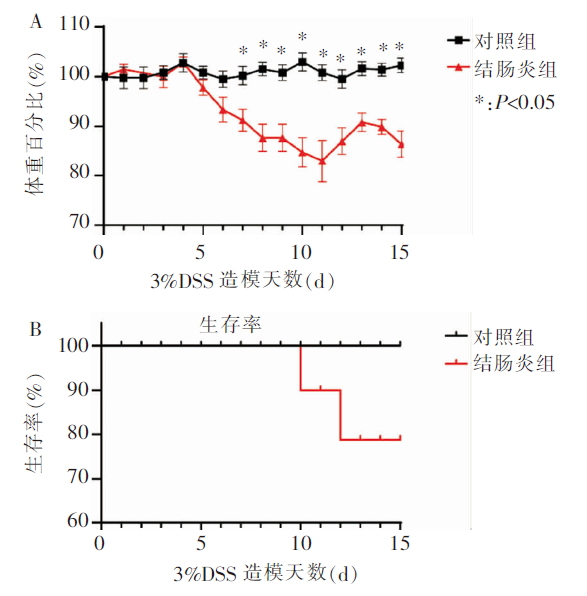
图2
2组小鼠的结肠上皮组织病理评分 A:结肠组织HE染色;B:结肠组织病理评分
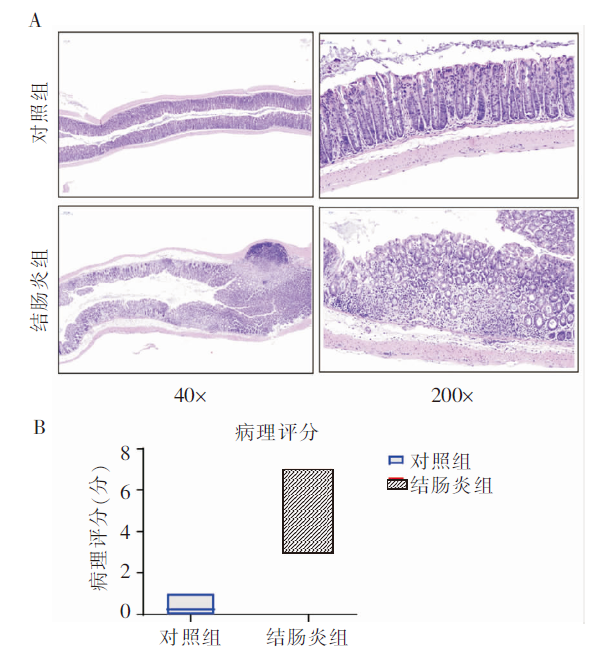
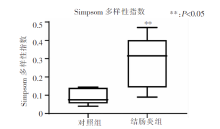
图3
2组小鼠肠道菌群Simpson值比较 注:Simpson指数值越大,群落多样性越低;盒子越窄,组内样本离散程度越小
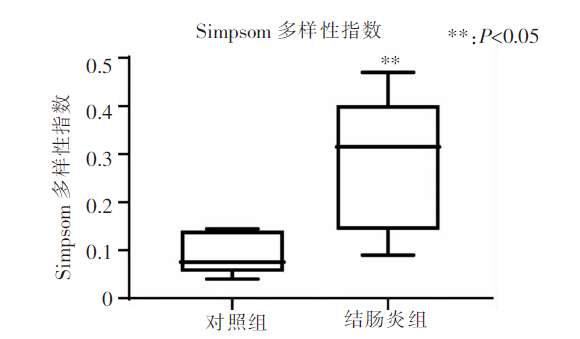
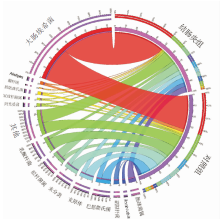
图4
肠炎小鼠肠道菌群分布改变情况 右边半圆代表2组菌群组成情况,左边半圆表示菌群在2组中的分布比例情况(仅显示分组中丰度最高的前15个菌群)
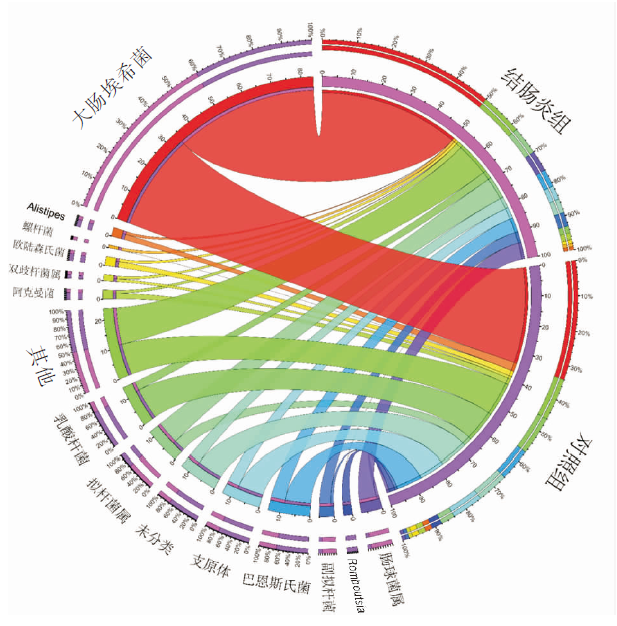
表2
2组小鼠间的菌群平均丰度差异分析
| 差异菌属 | 3%DSS组 平均丰度(%) | 对照组 平均丰度(%) | 均值差异 | P值 | 95.0%可信区间 下限 | 95.0%可信区间 上限 | 效应值 |
|---|---|---|---|---|---|---|---|
| 双歧杆菌属 | 0.235 67 | 1.799 96 | -1.564 29 | 0.040 65 | -3.031 42 | -0.097 16 | 0.424 62 |
| 肠球菌属 | 3.468 21 | 0.976 05 | 2.492 16 | 0.042 03 | 0.123 36 | 4.860 95 | 0.395 84 |
| 乳酸杆菌属 | 2.947 98 | 12.120 7 | -9.172 76 | 0.044 44 | -18.017 8 | -0.327 69 | 0.406 37 |
| 韦荣球菌属 | 0.902 27 | 0.133 94 | 0.7683 29 | 0.045 54 | 0.021 73 | 1.514 93 | 0.399 46 |
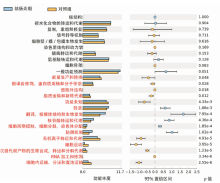
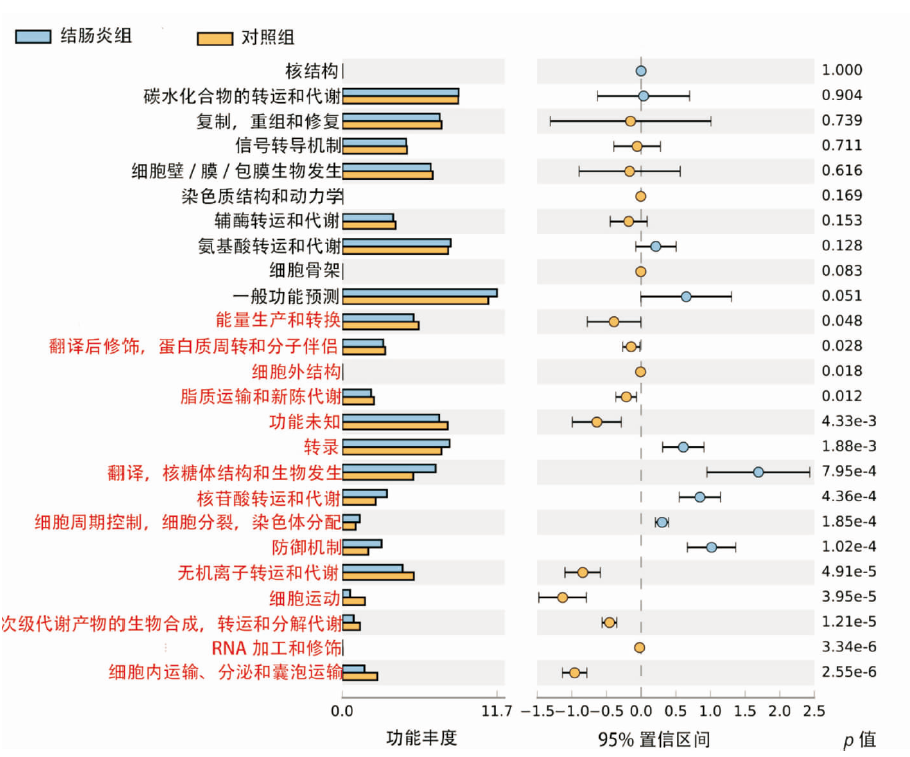
图5
16S rRNA测序分析2组小鼠肠道中功能基因丰度的差异 左侧为不同功能丰度在2组中的比例;中间为95%可信区间内,功能丰度丰度的差异比例;最右侧的值为P值,功能丰度差异显著用红色标识
| [1] | 张琴, 吴开春. 中国炎症性肠病癌变监测[J]. 医学新知, 2016, 26(4):235-237. |
| [2] |
Weisshof R, El Jurdi K, Zmeter N, et al. Emerging Thera-pies for Inflammatory Bowel Disease[J]. Adv Ther, 2018, 35(11):1746-1762.
doi: 10.1007/s12325-018-0795-9 pmid: 30374806 |
| [3] | 李军祥, 谭祥, 毛堂友. 溃疡性结肠炎中西医结合治疗策略[J]. 中国中西医结合杂志, 2017, 37(4):398-400. |
| [4] | de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy[J]. Nat Rev Gastroenterol Hepatol, 2017, 14(12):739-749. |
| [5] | 毛钰蕾, 周涛, 唐凌云, 等. 速激肽受体2对小鼠溃疡性结肠炎的影响[J]. 诊断学理论与实践, 2016, 15(6):578-581. |
| [6] | 郭晗, 张捷, 杨硕, 等. 肠道微生物与人类疾病关系的研究进展[J]. 检验医学, 2017, 32(12):1165-1172. |
| [7] |
Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways[J]. PLoS One, 2010, 5(1):e8578.
doi: 10.1371/journal.pone.0008578 URL |
| [8] |
Rosenbaum JT, Asquith MJ. The Microbiome: a Revolution in Treatment for Rheumatic Diseases?[J]. Curr Rheumatol Rep, 2016, 18(10):62.
doi: 10.1007/s11926-016-0614-8 pmid: 27641915 |
| [9] |
Knip M, Honkanen J. Modulation of type 1 diabetes risk by the intestinal microbiome[J]. Curr Diab Rep, 2017, 17(11):105.
doi: 10.1007/s11892-017-0933-9 URL |
| [10] |
Halfvarson J, Brislawn CJ, Lamendella R, et al. Dyna-mics of the human gut microbiome in inflammatory bowel disease[J]. Nat Microbiol, 2017, 2:17004.
doi: 10.1038/nmicrobiol.2017.4 pmid: 28191884 |
| [11] | Aleksandrova K, Romero-Mosquera B, Hernandez V. Diet, gut microbiome and epigenetics: emerging links with inflammatory bowel diseases and prospects for mana-gement and prevention[J]. Nutrients, 2017, 9(9),pii:E962. |
| [12] |
Lamas B, Richard ML, Leducq V et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands[J]. Nat Med, 2016, 22(6):598-605.
doi: 10.1038/nm.4102 URL |
| [13] |
Dheer R, Santaolalla R, Davies JM, et al. Intestinal epi-thelial toll-like receptor 4 signaling affects epithelial function and colonic microbiota and promotes a risk for transmissible colitis[J]. Infect Immun, 2016, 84(3):798-810.
doi: 10.1128/IAI.01374-15 URL |
| [14] |
Yang Y, Weng W, Peng J, et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-regu-lating Expression of MicroRNA-21[J]. Gastroenterology, 2017, 152(4):851-866.
doi: 10.1053/j.gastro.2016.11.018 URL |
| [15] |
de Weirdt R, Crabbé A, Roos S, et al. Glycerol supplementation enhances L. reuteri's protective effect against S. Typhimurium colonization in a 3-D model of colonic epithelium[J]. PLoS One, 2012, 7(5):e37116.
doi: 10.1371/journal.pone.0037116 URL |
| [16] | 余今菁, 李欢, 胡邱宇, 等. 基于高通量测序技术的溃疡性结肠炎患者肠道菌群多样性研究[J]. 华中科技大学学报(医学版), 2018, 47(4):460-465. |
| [17] | 姜洋, 赵秋枫, 王实, 等. 基于16S rRNA序列分析肠道菌群失调与溃疡性结肠炎的相关性[J]. 世界华人消化杂志, 2017, 25(36):3191-3202. |
| [18] |
Nunes NS, Kim S, Sundby M, et al. Temporal clinical, proteomic, histological and cellular immune responses of dextran sulfate sodium-induced acute colitis[J]. World J Gastroenterol, 2018, 24(38):4341-4355.
doi: 10.3748/wjg.v24.i38.4341 URL |
| [19] |
Kimura I, Ozawa K, Inoue D, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43[J]. Nat Commun, 2013, 4:1829.
doi: 10.1038/ncomms2852 pmid: 23652017 |
| [20] | 杜彩贺, 胡芳, 魏婷婷, 等. PCR-DGGE指纹图谱技术分析2型糖尿病模型小鼠胃微生物菌群结构[J]. 中国生物工程杂志, 2012, 32(3):25-31. |
| [21] |
Säemann MD, Böhmig GA, Osterreicher CH, et al. Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production[J]. FASEB J, 2000, 14(15):2380-2382.
pmid: 11024006 |
| [22] |
Lamas B, Richard ML, Leducq V, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands[J]. Nat Med, 2016, 22(6):598-605.
doi: 10.1038/nm.4102 URL |
| [23] |
Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disea-se[J]. Gut, 2004, 53(5):685-693.
doi: 10.1136/gut.2003.025403 pmid: 15082587 |
| [24] | 任科雨, 勇春明, 王艳婷, 等. 实验性结肠炎小鼠肠道菌群指纹图谱分析[J]. 胃肠病学和肝病学杂志, 2013, 22(9):894-897. |
| [25] |
Damman CJ, Miller SI, Surawicz CM, et al. The microbiome and inflammatory bowel disease: is there a therapeutic role for fecal microbiota transplantation?[J]. Am J Gastroenterol, 2012, 107(10):1452-1459.
doi: 10.1038/ajg.2012.93 URL |
| [26] |
Oberg TS, Ward RE, Steele JL, et al. Genetic and physiological responses of Bifidobacterium animalis subsp. lactis to hydrogen peroxide stress[J]. J Bacteriol, 2013, 195(16):3743-3751.
doi: 10.1128/JB.00279-13 URL |
| [27] |
LeBlanc JG, Milani C, de Giori GS, et al. Bacteria as vitamin suppliers to their host: a gut microbiota perspective[J]. Curr Opin Biotechnol, 2013, 24(2):160-168.
doi: 10.1016/j.copbio.2012.08.005 URL |
| [28] |
Aoki R, Kamikado K, Suda W, et al. A proliferative probiotic Bifidobacterium strain in the gut ameliorates progression of metabolic disorders via microbiota modulation and acetate elevation[J]. Sci Rep, 2017, 7:43522.
doi: 10.1038/srep43522 pmid: 28252037 |
| [29] |
Kołodziej M, Szajewska H. Lactobacillus reuteri DSM 17938 in the prevention of antibiotic-associated diarrhoea in children: protocol of a randomised controlled trial[J]. BMJ Open, 2017, 7(1):e013928.
doi: 10.1136/bmjopen-2016-013928 URL |
| [30] |
de Weirdt R, Crabbé A, Roos S, et al. Glycerol supplementation enhances L. reuteri's protective effect against S. Typhimurium colonization in a 3-D model of colonic epithelium[J]. PLoS One, 2012, 7(5):e37116.
doi: 10.1371/journal.pone.0037116 URL |
| [31] |
Bertin Y, Habouzit C, Dunière L, et al. Lactobacillus reuteri suppresses E. coli O157:H7 in bovine ruminal fluid: Toward a pre-slaughter strategy to improve food safety?[J]. PLoS One, 2017, 12(11):e0187229.
doi: 10.1371/journal.pone.0187229 URL |
| [32] | 彭玄杰. 双歧杆菌三联活菌联合美沙拉嗪治疗溃疡性结肠炎的临床疗效分析[J]. 中国中西医结合消化杂志, 2014, 22(11):672-673. |
| [33] | 梁涛. 乳酸乳杆菌对溃疡性结肠炎患者辅助治疗作用及相关机制研究[J]. 中国医院药学杂志, 2015, 35(11):1018-1022. |
| [34] |
Ihara S, Hirata Y, Koike K. TGF-β in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota[J]. J Gastroenterol, 2017, 52(7):777-787.
doi: 10.1007/s00535-017-1350-1 URL |
| [35] |
Ramakrishnan SK, Zhang H, Ma X, et al. Intestinal non-canonical NFκB signaling shapes the local and systemic immune response[J]. Nat Commun, 2019, 10(1):660.
doi: 10.1038/s41467-019-08581-8 pmid: 30737385 |
| [36] |
Lavoie S, Conway KL, Lassen KG, et al. The Crohn's disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response[J]. Elife, 2019, 8(pii):e39982.
doi: 10.7554/eLife.39982 URL |
| [37] |
Shen W, Shen M, Zhao X, et al. Anti-obesity Effect of Capsaicin in Mice Fed with High-Fat Diet Is Associated with an Increase in Population of the Gut Bacterium Akkermansia muciniphila[J]. Front Microbiol, 2017, 8:272.
doi: 10.3389/fmicb.2017.00272 pmid: 28280490 |
| [38] |
Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome[J]. Science, 2006, 312(5778):1355-1359.
doi: 10.1126/science.1124234 URL |
| [39] |
Ahmed J, Reddy BS, Mølbak L, et al. Impact of probio-tics on colonic microflora in patients with colitis: a prospective double blind randomised crossover study[J]. Int J Surg, 2013, 11(10):1131-1136.
doi: 10.1016/j.ijsu.2013.08.019 URL |
| [40] | 陈迪, 陈敏, 张玉洁, 等. 疑似溃疡性结肠炎的药物诱导性肠炎[J]. 中华消化杂志, 2017, 37(3):197-200. |
| [1] | 许飞, 尹明月, 王伟, 董治亚, 陆文丽, 余熠, 王歆琼, 王俊祺, 肖园. 性早熟女童肠道菌群和抗生素耐药性的宏基因组分析[J]. 诊断学理论与实践, 2022, 21(01): 52-61. |
| [2] | 李林, 安静静, 王俊祺, 王歆琼, 董治亚. 16S rRNA第二代测序技术分析特发性身材矮小儿童肠道菌群构成的特征及相关发病机制研究[J]. 诊断学理论与实践, 2021, 20(02): 149-154. |
| [3] | 李惠, 冯洁, 韩立中. 高通量测序技术分析无特定病原体级实验小鼠肠道的菌群组成[J]. 诊断学理论与实践, 2020, 19(1): 55-62. |
| [4] | 安静静, 王俊祺, 肖园, 陆文丽, 李林, 王伟, 董治亚. 16S rRNA高通量测序分析肠道菌群对小于胎龄大鼠生长追赶的影响及其可能的机制[J]. 诊断学理论与实践, 2020, 19(04): 375-380. |
| [5] | 陈瑶瑶, 顾爱华. 氧化三甲胺与心血管疾病关系的研究进展[J]. 诊断学理论与实践, 2019, 18(2): 237-240. |
| [6] | 毛钰蕾, 周涛, 唐凌云, 张洪信, 王铸钢. 速激肽受体2对小鼠溃疡性结肠炎的影响[J]. 诊断学理论与实践, 2016, 15(06): 578-581. |
| [7] | 方一, 刘倩, 钟捷, 龚彪, 夏璐,. 巨噬细胞极化对溃疡性结肠炎病情发展的影响[J]. 诊断学理论与实践, 2015, 14(06): 568-572. |
| [8] | 方一, 刘倩, 陈璐, 卞兆连, 苗琦, 马雄, 夏璐,. 淋巴毒素α1/β2及其受体在溃疡性结肠炎肠道黏膜免疫中的作用及意义[J]. 诊断学理论与实践, 2015, 14(03): 223-228. |
| [9] | 刘倩, 方一, 卞兆连, 苗琦, 戴欣, 陈璐, 马雄, 夏璐,. C型凝集素样受体Dectin-1在溃疡性结肠炎肠道黏膜免疫中的作用[J]. 诊断学理论与实践, 2014, 13(06): 579-583. |
| [10] | 夏璐, 刘慧黎, 戴欣, 徐凯, 诸琦,. 应用小探头超声预测溃疡性结肠炎缓解的价值[J]. 诊断学理论与实践, 2008, 7(06): 605-608. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||