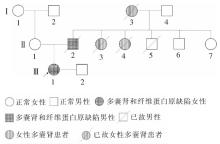
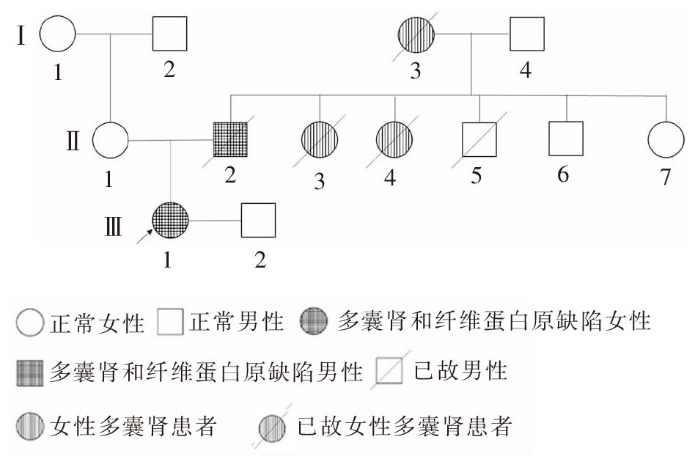
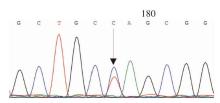
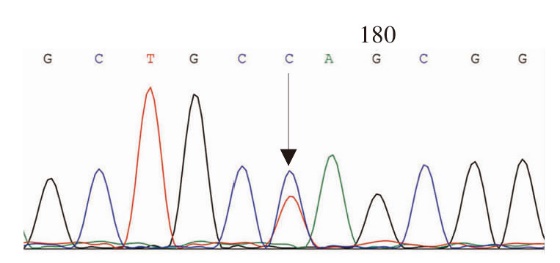
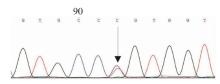
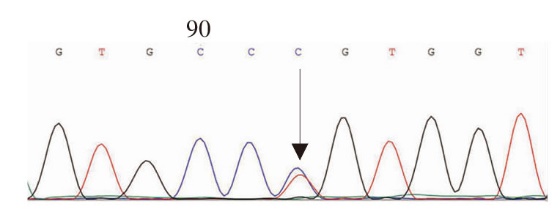
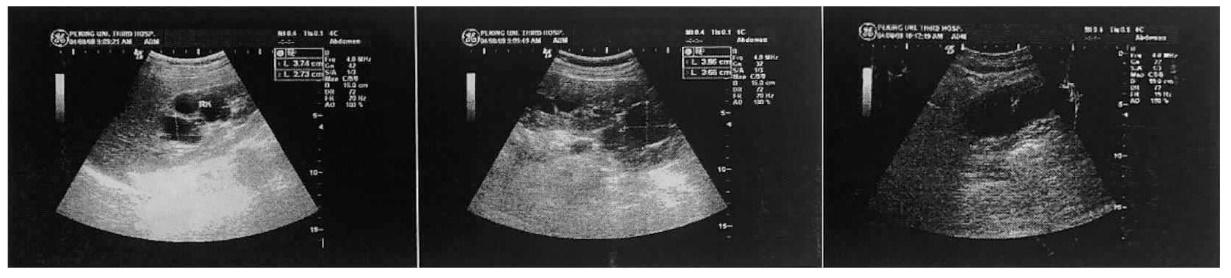
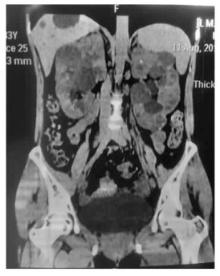
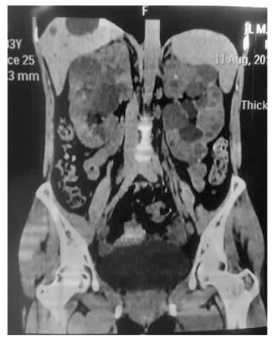
Journal of Internal Medicine Concepts & Practice ›› 2023, Vol. 18 ›› Issue (05): 328-333.doi: 10.16138/j.1673-6087.2023.05.004
• Original article • Previous Articles Next Articles
ZHOU Liyanga, ZHANG Chunlib, DING Qiulana, LI Yab( )
)
Received:2023-03-09
Online:2023-10-30
Published:2024-01-25
Contact:
LI Ya
E-mail:yayayaly@126.com
CLC Number:
ZHOU Liyang, ZHANG Chunli, DING Qiulan, LI Ya. Genetic diagnosis and clinical analysis of congenital dysfibrinogenemia with polycystic disease: a case report and literature review[J]. Journal of Internal Medicine Concepts & Practice, 2023, 18(05): 328-333.
| [1] |
Dell KM, Matheson M, Hartung EA, et al. Kidney disease progression in autosomal recessive polycystic kidney disease[J]. J Pediatr, 2016, 171: 196-201.
doi: 10.1016/j.jpeds.2015.12.079 URL |
| [2] |
Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease[J]. Nat Rev Dis Primers, 2018, 4(1):50.
doi: 10.1038/s41572-018-0047-y pmid: 30523303 |
| [3] | Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant[DB/OL]. 2022. https://www.ncbi.nlm.nih.gov/books/NBK1246/. |
| [4] |
Lanktree MB, Chapman AB. New treatment paradigms for ADPKD: moving towards precision medicine[J]. Nat Rev Nephrol, 2017, 13(12): 750-768.
doi: 10.1038/nrneph.2017.127 pmid: 28989174 |
| [5] |
Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease[J]. Lancet, 2019, 393(10174): 919-935.
doi: S0140-6736(18)32782-X pmid: 30819518 |
| [6] |
Neerman-Arbez M, de Moerloose P, Casini A. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders[J]. Semin Thromb Hemost, 2016, 42(4): 356-365.
doi: 10.1055/s-0036-1571340 pmid: 27019463 |
| [7] |
Richard M, Celeny D, Neerman-Arbez M. Mutations accounting for congenital fibrinogen disorders[J]. Semin Thromb Hemost, 2022, 48(8): 889-903.
doi: 10.1055/s-0041-1742170 URL |
| [8] |
Casini A, Neerman-Arbez M, Ariëns RA, et al. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management[J]. J Thromb Haemost, 2015, 13(6): 909-919.
doi: 10.1111/jth.12916 pmid: 25816717 |
| [9] |
Casini A, Blondon M, Lebreton A, et al. Natural history of patients with congenital dysfibrinogenemia[J]. Blood, 2015, 125(3): 553-561.
doi: 10.1182/blood-2014-06-582866 pmid: 25320241 |
| [10] |
Mangolini A, de Stephanis L, Aguiari G. Role of calcium in polycystic kidney disease[J]. World J Nephrol, 2016, 5(1): 76-83.
doi: 10.5527/wjn.v5.i1.76 URL |
| [11] |
Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease[J]. J Clin Invest, 2014, 124(6): 2315-2324.
doi: 10.1172/JCI72272 pmid: 24892705 |
| [12] |
Bergmann C. Recent advances in the molecular diagnosis of polycystic kidney disease[J]. Expert Rev Mol Diagn, 2017, 17(12): 1037-1054.
doi: 10.1080/14737159.2017.1386099 pmid: 28952822 |
| [13] |
Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD)[J]. Kidney Int, 2015, 88(1): 17-27.
doi: 10.1038/ki.2015.59 pmid: 25786098 |
| [14] |
Obeidova L, Elisakova V, Stekrova J, et al. Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease[J]. BMC Med Genet, 2014, 15: 41.
doi: 10.1186/1471-2350-15-41 pmid: 24694054 |
| [15] | Trujillano D, Bullich G, Ossowski S, et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing[J]. Mol Genet Genomic, 2014, 2(5): 412-421. |
| [16] |
Rahbari-Oskoui F, Williams O, Chapman A. Mechanisms and management of hypertension in autosomal dominant polycystic kidney disease[J]. Nephrol Dial Transplant, 2014, 29(12): 2194-2201.
doi: 10.1093/ndt/gft513 URL |
| [17] | Nauli SM, Jin X, Hierck BP. The mechanosensory role of primary cilia in vascular hypertension[J]. Int J Vasc Med, 2011: 376281. |
| [18] |
Kocyigit I, Eroglu E, Gungor O. Clinical problems in hemodialysis patients with autosomal dominant polycystic kidney disease[J]. Semin Dial, 2018, 31(3): 268-277.
doi: 10.1111/sdi.2018.31.issue-3 URL |
| [19] |
Rozenfeld MN, Ansari SA, Shaibani A, et al. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms?[J]. AJNR Am J Neuroradiol, 2014, 35(1): 3-9.
doi: 10.3174/ajnr.A3437 URL |
| [20] |
Perrone RD, Malek AM, Watnick T. Vascular complications in autosomal dominant polycystic kidney disease[J]. Nat Rev Nephrol, 2015, 11(10): 589-598.
doi: 10.1038/nrneph.2015.128 pmid: 26260542 |
| [21] |
Etminan N, Rinkel GJ. Unruptured intracranial aneurysms: development, rupture and preventive management[J]. Nat Rev Neurol, 2016, 12(12): 699-713.
doi: 10.1038/nrneurol.2016.150 pmid: 27808265 |
| [22] |
Cagnazzo F, Gambacciani C, Morganti R, et al. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management[J]. Acta Neurochir (Wien), 2017, 159(5): 811-821.
doi: 10.1007/s00701-017-3142-z URL |
| [23] |
Qian Q, Younge BR, Torres VE. Retinal arterial and venous occlusions in patients with ADPKD[J]. Nephrol Dial Transplant, 2007, 22(6): 1769-1771.
doi: 10.1093/ndt/gfm034 URL |
| [24] | Kuo IY, Chapman AB. Polycystins, ADPKD, and cardiovascular disease[J]. Kidney Int Rep, 2019, 5(4): 396-406. |
| [25] |
Gabow PA, Duley I, Johnson AM. Clinical profiles of gross hematuria in autosomal dominant polycystic kidney disease[J]. Am J Kidney Dis, 1992, 20(2): 140-143.
doi: 10.1016/s0272-6386(12)80541-5 pmid: 1496966 |
| [26] |
Colbert GB, Elrggal ME, Gaur L, et al. Update and review of adult polycystic kidney disease[J]. Dis Mon, 2020, 66(5): 100887.
doi: 10.1016/j.disamonth.2019.100887 URL |
| [27] |
Nishiura JL, Neves RF, Eloi SR, et al. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients[J]. Clin J Am Soc Nephrol, 2009, 4(4): 838-844.
doi: 10.2215/CJN.03100608 URL |
| [28] |
Torres VE, Wilson DM, Hattery RR, et al. Renal stone disease in autosomal dominant polycystic kidney disease[J]. Am J Kidney Dis, 1993, 22(4): 513-519.
pmid: 8213789 |
| [29] |
Gambaro G, Fabris A, Puliatta D, et al. Lithiasis in cystic kidney disease and malformations of the urinary tract[J]. Urol Res, 2006, 34(2): 102-107.
doi: 10.1007/s00240-005-0019-z pmid: 16416113 |
| [30] |
Moist LM, Churchill DN, House AA, et al. Regular monitoring of access flow compared with monitoring of venous pressure fails to improve graft survival[J]. J Am Soc Nephrol, 2003, 14(10): 2645-2653.
pmid: 14514744 |
| [31] |
Schwab SJ, Oliver MJ, Suhocki P, et al. Hemodialysis arteriovenous access: detection of stenosis and response to treatment by vascular access blood flow[J]. Kidney Int, 2001, 59(1): 358-362.
pmid: 11135091 |
| [32] |
Hsieh MY, Cheng CH, Chen CH, et al. The association of long-term blood pressure variability with hemodialysis access thrombosis[J]. Front Cardiovasc Med, 2022, 9:881454.
doi: 10.3389/fcvm.2022.881454 URL |
| [33] |
Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia[J]. Thromb Haemost, 1995, 73(1): 151-161.
doi: 10.1055/s-0038-1653741 URL |
| [34] | Casini A, How I treat dysfibrinogenemia[J]. Blood, 2021, 138(21): 2021-2030. |
| [35] |
Vakalopoulou S, Mille-Baker B, Mumford A, et al. Fibrinogen Bbeta14 Arg—>Cys: further evidence for a role in thrombosis[J]. Blood Coagul Fibrinolysis, 1999, 10(7): 403-408.
doi: 10.1097/00001721-199910000-00001 URL |
| [1] |
ZHAO Yifei, ZOU Yun, CHEN Hui, et al.
Analysis of mutation type and clinical significance of congenital melanocytic nevi [J]. Journal of Tissue Engineering and Reconstructive Surgery, 2023, 19(3): 258-. |
| [2] | YANG Cuiyan, WANG Haoyu, CHEN Xiaosong, SHEN Kunwei. Study on tumour suppressor gene TP53 mutation and prognosis in patients with triple-negative breast cancer [J]. Journal of Surgery Concepts & Practice, 2022, 27(05): 421-428. |
| [3] | HAO Xu, WANG Weiming. Fabry disease presenting with renal disease as the main manifestation diagnosed by renal biopsy: a case report [J]. Journal of Diagnostics Concepts & Practice, 2022, 21(04): 527-529. |
| [4] | ZHAO Yingmei, NIE Hongming, ZHANG Jue, HUANG Yan. Detection of five coagulation indices in patients with cirrhosis and its clinical significance [J]. Journal of Diagnostics Concepts & Practice, 2021, 20(05): 480-483. |
| [5] | LEI Hang, FAN Liangfeng, CAI Xiaohong, WANG Yuqing, LIU Xi, JIN Sha, SHEN Wei, LU Qiong, XIANG Dong, WANG Xuefeng, ZOU Wei. The study on molecular basis of ABO blood subgroups in the Chinese population [J]. Journal of Diagnostics Concepts & Practice, 2020, 19(04): 364-369. |
| [6] | . [J]. Journal of Internal Medicine Concepts & Practice, 2020, 15(02): 94-98. |
| [7] | PENG Zhenping, XIANG Xixi, ZHANG Sujiang, LI Jiaming. Chronic neutrophilic leukemia with leukemia-like reaction as the first-onset manifestation: a report of 2 cases and literature review [J]. Journal of Diagnostics Concepts & Practice, 2020, 19(02): 122-128. |
| [8] | PENG Meifang,WU Yingzhi,CHEN Jieyi,JIN Li,MU Xiongzheng,WANG Sijia. Differences in the Clinical Phenotypes of Apert Syndrome with the FGFR2 Mutation Site Ser252Trp and Pro253Arg: Based on Meta Analysis [J]. Journal of Tissue Engineering and Reconstructive Surgery, 2018, 14(2): 67-72. |
| [9] | . [J]. Machine Design & Research, 2018, 34(06): 41-46. |
| [10] | XU Mengyang, HE Bin, HUA Baolai, JI Wei, SHEN Lianjun, SHI Qingqing, SUN Xing, YU Ju, XIE Xiaoyan, SUN Mei, GU Jian. The discrepancy of thrombin time results between two kinds of thrombolyzer in a patient associated with heparin like anticoagulant [J]. Journal of Diagnostics Concepts & Practice, 2018, 17(06): 707-710. |
| [11] | CAI Rong, MIN Xuewen, CHEN Meirong, SHEN Yating, SHI Qunli, ZHOU Xiaodie. Expression of BRAF V600E (VE1) in thyroid papillary carcinoma and its clinical significance [J]. Journal of Diagnostics Concepts & Practice, 2018, 17(05): 552-556. |
| [12] | WANG Dengfeng, CUI Wenyan, ZOU Wei, LI Fang, WANG Xuefeng, CAI Xiaohong. Molecular mechanism of Ax subtype caused by p.M142I mutation in alpha 1-3-N-acetylgalactosaminyltransferase [J]. Journal of Diagnostics Concepts & Practice, 2018, 17(03): 260-265. |
| [13] | LU Jing, XU Yufei, QING Yanrong, HAN Cong, LI Niu, YU Tingting, YAO Ruen, WANG Jian. Concurrent gene mutation analysis of a developmental delayed child with Rett syndrome and Noonan syndrome [J]. Journal of Diagnostics Concepts & Practice, 2018, 17(02): 147-150. |
| [14] | JIN Peipei, LIANG Qian, DAI Jing, DING Qiulan, SUN Shunchang, WANG Xuefeng. Phenotype and genotype analysis of a Chinese pedigree with 2N type von Willebrand disease [J]. Journal of Diagnostics Concepts & Practice, 2018, 17(02): 151-154. |
| [15] | SHAN Yuhua,CHU Jun,HU Ming,XU Min,CHEN Qimin. Feasibility Analysis of Sirolimus Treatment for KMS in Young Children [J]. Journal of Tissue Engineering and Reconstructive Surgery, 2017, 13(5): 244-247. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||