
Highlight
• We utilize a pre-trained model to enrich the information of sequence embedding, and self-attention mechanism to capture global dependencies.
• Our inhibition predictor achieves the best performance on both accuracy and generalization.
• We construct a simple and user-friendly approach to design both potent and specific siRNAs.
Introduction
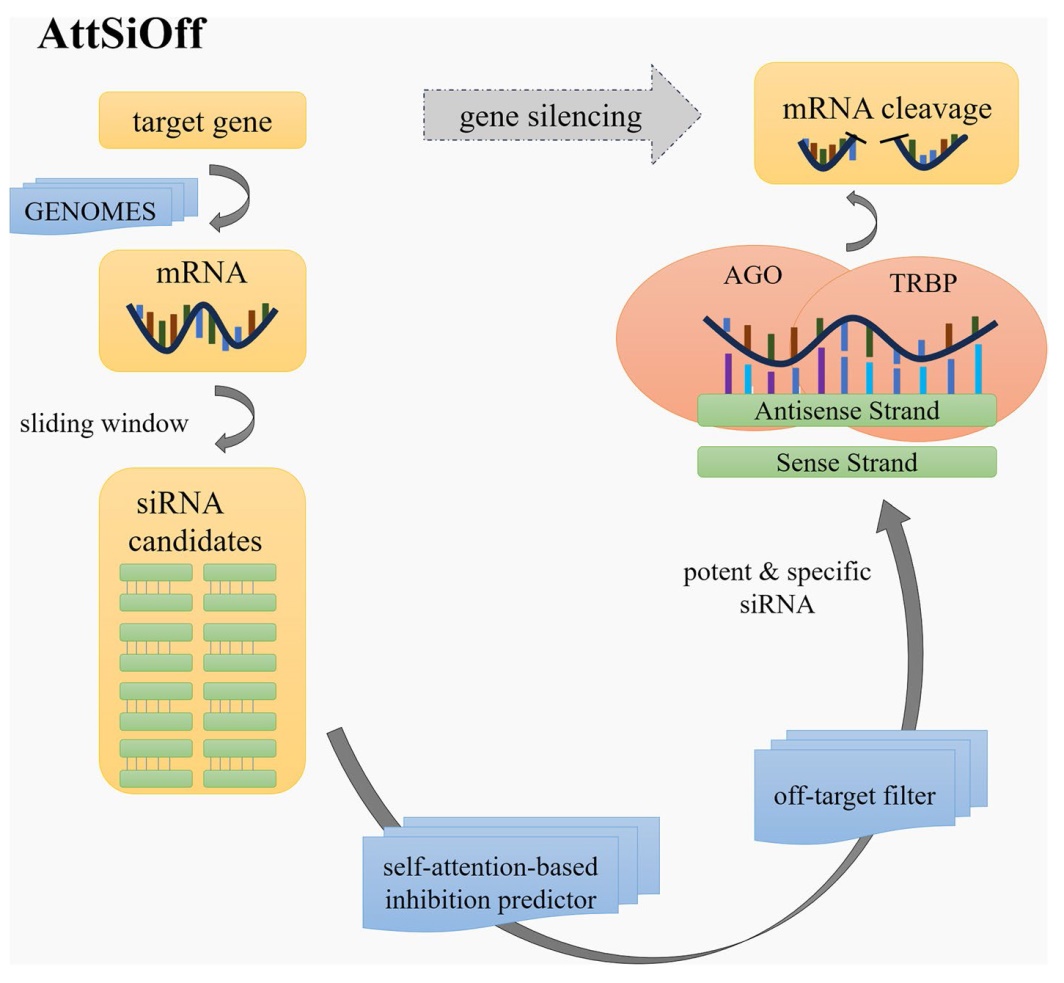
RNA interference (RNAi), also known as post-transcriptional gene silencing, can resist parasitic and pathogenic nucleic acids, and regulate specific gene expression. It has been developed into a mature technology to mediate gene expression manually and probe gene function [1]. RNAibased regulators include 21 ~ 23-nucleotide small interfering RNA (siRNA) and ~ 22-nucleotide microRNA (miRNA). In this paper, we mainly discuss the function and prediction of siRNA. With the help of Argonaute Family Protein 2 (AGO2) and TAR RNA Binding Proteins (TRBP), the antisense strand (AS) in the siRNA duplex will bind with the target mRNA by Watson-Crick base pairing. If the entire AS can hybridize with the target mRNA, it will introduce mRNA cleavage to prohibit the translation process. If only the seed region of AS hybridizes with the target mRNA, it will induce mRNA degradation and repress the translation process [2⇓⇓-5].
Usually, target mRNA is composed of hundreds or thousands of nucleotides, from which we can generate a massive amount of siRNA candidates by the sliding window method. However, the knockdown efficiency of siRNA, also called inhibition, may vary a lot with a slight change in its composition [6]. The silencing inhibition is mainly determined by the sequence patterns, binding affinity, and the secondary structure around the binding regions, while the specificity is mostly determined by off-target effects [7]. Compared with off-target effects, inhibition is more difficult to predict. All the time, researchers have been focusing on the challenges to predict the inhibition of siRNA accurately.
Early on, methods are mainly developed from rather small datasets, which often contain biased information. For example, Amarzguiouti et al. performed a statistical analysis of only 46 siRNAs to identify the preferences of nucleotides on each position of the siRNA duplex. They find that the motifs U1 or G19 are strongly related to poor inhibition [8].
With the growth of valid samples, algorithms based on machine learning have been developed in a data-driven way. Heusken et al. make the greatest contribution to enriching the relevant dataset. They collected 2431 siRNAs targeting 34 mRNAs with corresponding validated inhibitions. They developed a model named BIOPREDsi using the Stuttgart Neural Net Simulator to achieve a rather high Pearson Correlation Coefficient (PCC) of 0.66 [9]. Vert et al. perform a LASSO-based regression model, to conveniently estimate the importance of each feature. They use the preference for specific nucleotides on some positions, as well as short asymmetric base motifs, as the input [10]. Ichihara et al. developed a simple linear regression algorithm, i-Score, which is only comprised of nucleotide preferences at each position as the input, and achieves a comparative PCC with s-Biopredsi [11]. However, these methods still suffer from the incompetence of detecting hidden features.
In recent years, Convolutional Neural Network (CNN) has been successfully applied to diverse fields, such as machine translation, object detection, protein interaction, etc. It has also been used in siRNA inhibition prediction and showed remarkable enhancement of precision. Similar to TextCNN, the work done by Han et al. utilizes multiple convolutional kernels to detect unknown but helpful motifs from local target mRNA sequences, preprocessed by one-hot encoding. Then it uses average pooling and maximum pooling layers to extract the most representative features. The thermodynamic property calculated from AS is concatenated with the pooling output and then normalized in batch. At last, a neural network with one hidden layer of 25 nodes is applied, and this model generates the prediction score via the sigmoid activation function [12]. It achieves a remarkable improvement in PCC compared with traditional models. However, the inadequate input features and simply hidden layers limit its performance. The forceful pooling operations result in a great loss of information.
Aside from CNN, Graph Neural Network (GNN) is another common deep learning algorithm used in bioinformatics. Biological molecules are regarded as nodes, and their relationships can be represented with edges connecting different nodes [13]. The graph is an intrinsically good structure to model topology and capture hidden interrelationships in non-structural data. Massimo et al. propose a GNN-based model for siRNA inhibition prediction for the first time. There are three types of nodes in their graph. The first is a siRNA node, with 3-mer counting as its feature. The second is the target mRNA node, with 4-mer counting as its feature. The third is the siRNA-mRNA interaction node, with the thermodynamic parameters calculated from Gibbs energy and RNAup program as its features. Here, they replace the interaction edge with a node, for the sake of thoroughly incorporating the interaction information into the graph. They consider the inhibition as the property of the siRNA-mRNA interaction node to predict. This algorithm achieves a better performance than the aforementioned CNN-based method. However, the k-mer counting features are still insufficient to represent the characteristics of siRNA or mRNA sequences. The graph is fixed with nodes from the trainset and test set, resulting in its disability to predict the inhibition of new siRNA samples [14].
Despite that various research have been developed to predict siRNA inhibition, there is still room for developing a new algorithm with better accuracy and generalization. This can be achieved by optimizing feature selection and modeling process. For example, existing methods usually use one-hot encoding to transform siRNA sequence into binary sparse matrix. It lacks inter-relationship among different kinds of nucleotides. To extract more information hidden in sequence, Chen et al. propose a transformer-based model, named RNA Foundation Model (RNA-FM). It is trained on 23 million ncRNA sequences via self-supervised learning. The clustering results indicate that the pre-trained RNA-FM embedding contains much sequential and functional patterns [15]. This model is a good way to encode RNA sequence in our research. As for model structure, the transformer used in RNA-FM model is based on self-attention mechanism. It enables the model to measure the importance of different elements in the input sequence and dynamically adjust their impact on the output. Compared with recurrence and convolutions, self-attention can capture long-distance dependencies more efficiently and precisely, when applied to long sentences[16]. And DNA or RNA sequence is just composed of a lot of nucleotides.
However, some challenges prevent siRNA from being applied into clinical trials, such as limited longevity and inevitable off-target effects. [17⇓-19]. Off-target effects will result in serious misjudgment of inhibition. And silencing uncertain mRNAs may negatively interfere with some significant biochemical pathways. Compared with difficult inhibition prediction, the off-target effect is easier to analyze with some definite criteria.
In this study, we propose a novel approach for siRNA inhibition and off-target effect prediction, named AttSi- Off. This self-attention-based inhibition predictor employs two types of features. One is the embedding of siRNA and local target mRNA sequences, generated from a pre-trained RNA-FM model. The other is the prior-knowledge-based characteristics of AS, including the thermodynamic parameters, the secondary structure, GC content, PSSM score, etc. This predictor is comprised of two parts: a feature extraction module and a fully connected module. In the feature extraction module, the multi-head self-attention mechanism is used to further extract hidden information by constructing the interaction of every nucleotide with others. To the best of our knowledge, it is the first time of self-attention mechanism has been used in the prediction of siRNA inhibition. In the fully connected module, the high-dimensional representations produced by multi-head self-attention are concatenated with other features and go through a deep and wide neural network, to produce a prediction score. The tenfold cross-validation results and cross-dataset experiments both show that our predictor achieves state-of-the-art performance, compared with other existing methods.
To facilitate and streamline siRNA design, we combine the predictor with an mRNA searching package and an off-target filter. The mRNA searching package can find all mature mRNAs for any given gene name. The off-target filter can calculate the amount of possible unwanted offtarget binding sites, which affects the specificity of siRNA. We testify to the practicability and maneuverability of our pipeline on five siRNA drugs from Alnylam Company. The results show its great simplicity and effectiveness.
We proclaim the following contributions in our approach: (1) We apply the pre-trained RNA-FM model to greatly enrich the embedding of the RNA sequences, instead of using the classic one-hot binary encoding method. (2) We successfully employ a self-attention mechanism in the work of siRNA inhibition prediction for the first time to capture the global and local dependencies. (3) Our predictor achieves the best performance on both prediction accuracy and cross-dataset generalization, compared with other methods. (4) We construct a simple and user-friendly approach to automatically design both potent and specific siRNAs.
Materials and methods
Datasets
As suggested [12], we obtain as many experimentally validated siRNAs as possible. In this study, 3536 siRNAs from the work done by Huesken [9], Reynolds [20], Vickers [21], Haborth [22], Takayuki [11], and Ui-Tei [23], are collected. We divide these samples into three datasets according to their experimental conditions, namely DH, DR, and DT. The detailed composition of these three datasets is shown in Table 1.
Table 1 The detailed composition of three siRNA datasets. It shows the number of siRNAs and target mRNAs in each dataset, as well as corresponding publishers. Two siRNAs are removed in DT due to the limitation of i-score website, and two are removed in DR as a result of lacking binding sites on reported mRNAs |
 |
Two miRNAs in DR are removed as a result of failing to find binding sites on reported target mRNAs. Two siRNAs in DT are removed due to the limitation of the i-Score website, which will be explained later. In addition, the inhibition labels of these three datasets range from 0 to 134.1, -27.8 to 98.9, and 0 to 97, respectively. To unify the data distribution, they are normalized individually before being combined as a DHRT dataset.
Apart from the public datasets, we collect five siRNA medicines from Alnylam Company, which have been applied to clinical and diagnostic usage in recent years (Supplementary Table S1). Although these siRNAs are chemically modified to strengthen the potency, prolong the longevity, and weaken off-target effects, we can remove the chemical components here and use their original sequences to further validate the robustness and generalization of our inhibition predictor, as well as the practicability and maneuverability of our siRNA design tool.
The architecture of inhibition predictor
As is shown in Fig. 1, our inhibition predictor consists of a feature extraction module, a self-attention module, and a fully connected module.

Fig. 1 The architecture of our self-attention-based siRNA inhibition predictor. It is composed of three modules: feature extraction module, selfattention module, and fully connected module |
Feature extraction module
In this module, we mainly extract two types of features. One is siRNA and local target mRNA sequence contexts, generated from the pre-trained RNA-FM model. The other is the prior knowledge-based characteristics of AS, including thermodynamic parameters, k-mer counting, PSSM score, the secondary structure, and GC content.
For the sequence contexts, we displace classic one-hot encoding with pre-trained RNA-FM embedding. Generally, only the core region (19 nucleotides from the 5' end) of AS will hybridize with the target mRNA. Here, the local target mRNA sequence is defined as 19 nucleotides on the binding site plus 20 downstream and 20 upstream nucleotides (59 nucleotides total), as suggested in the work of Han et al. [12]. RNA-FM model will transform each nucleotide into a 640-dimensional continuous vector, which proves to contain structure-related and function-related information. The 21-nucleotide AS and 59-nucleotide local target mRNA sequences are transformed into feature matrixes with sizes of 21 × 640, and 59 × 640, respectively.
Note that not all local target mRNA has complete flanking regions around its binding site, so we use a special 640-dimensional vector [0.05, 0.05…0.05] to represent the missing nucleotide during embedding.
For the prior knowledge-based characteristics of AS, we mainly consider five groups of relevant features. Although they are described in previous research from biased and small datasets, they do play a role in predicting the inhibition of siRNA.
The first one is thermodynamic stability, which has an importance on inhibition and longevity, as well as determining which strand in siRNA duplex will hybridize with target mRNA [2,11,24,25]. We use the Gibbs free energy to describe the thermodynamic characteristics of the core region of AS. For instance, if the two adjacent bases are A and G, then the energy is -2.08 kJ/mol, according to Supplementary Table S2. There are 20 parameters in total, including 18 features calculated from every two adjacent bases, 1 feature from the energy difference between its 5' end and 3' end, and 1 feature of overall energy.
The second one is k-mer counting. We count the presences of 1-mer (4 motifs), 2-mer (16 motifs), and 3-mer (64 motifs) segments in AS. There are 84 parameters in total.
The third one is the PSSM score. Position Specific Scoring Matrix (PSSM) describes the possibility of observing every kind of nucleotide on each position. Here, we generate the PSSM from statistical analysis of the entire dataset. For any siRNA to predict, we utilize the fixed PSSM to estimate the preference for specific nucleotides in some positions. There is only 1 parameter for this feature.
The fourth one is related to secondary structure. Unstructured AS can mediate more active gene silencing. That means folded antisense strand is hard to hybridize with target mRNA, consequently reducing its inhibition [26]. Here we use the RNAfold program to calculate the minimum free energy and possible base pairing percentage as the secondary- structure-related features [27]. There are 3 parameters in all.
The last one is GC content, which affects the inhibition by changing the thermodynamic property. The stability between bases G and C is much stronger than that between bases A and U, as is shown in Supplementary Table S2. High GC content will result in the difficulty of SS and AS to separate from each other, while low GC content will lead to the instability of the hybridization of AS and target mRNA. Thus, a moderate GC content is much preferable. Besides, we calculate the maximum length of continuous base G or C, which represents the lower bound of the stability of the entire sequence. There are 2 parameters for this type of feature.
In summary, we normalize the above five groups of features individually to uniform the input distribution. And the prior-knowledge-based characteristics of AS form a feature matrix with a size of 1 × 110.
Self-attention module
The self-attention module is used to extract hidden features in AS and local target mRNA sequence embedding, after adding the sine and cosine position embedding.
The submodule for AS is composed of one linear layer, one TransformerEncoder (d_model = 16, num_layers = 4, nhead = 2), another linear layer, and one flattened layer. The initial dimension of AS embedding (21 × 640) produced by RNA-FM is too big to extract valid information. It may contain many redundant and task-irrelevant features, and the high-dimensional input will overburden the following selfattention mechanism. Therefore, the first linear layer is used to squeeze it to form a 16-dimensional high-level representation (21 × 16), to reduce the computation complexity. The following TransformerEncoder will keep the same dimension during forward propagation. To concatenate with other features, the second linear layer will sequence the 16-dimensional self-attention output to form a lower 4-dimensional representation (21 × 4), and the flattened layer will transform it into a 1-dimensional output with the size of 1 × 84. The second linear layer plays the similar role of pooling, but it will take all input features into account, instead of just selecting the maximum or average value.
The submodule for mRNA sequence is composed of one linear layer, one TransformerEncoder (d_model = 8, num_ layers = 2, nhead = 1), and another linear layer. Similarly, the first linear layer will squeeze the initial RNA-FM embedding to form an 8-dimensional high-level representation (59 × 8), and the second linear layer will transform the self-attention output into a 1-dimensional feature matrix with a size of 1 × 59 directly. We think the information in AS embedding is more useful and decisive than that in mRNA embedding, and that is why we keep fewer parameters in the submodule for mRNA.
In summary, the self-attention module will generate highlevel representations for AS with the size of 1 × 84, and for local target mRNA with the size of 1 × 59.
Fully connected module
The fully connected module is composed of a batch normalization layer, four non-linear sublayers, and a sigmoid activation layer. Each non-linear sublayer consists of a linear layer, a ReLU activation function, and a dropout layer. Input features are made up of the output of the self-attention module and other prior-knowledge-based features, the size of which is 1 × 253 (84 + 59 + 110). We normalize the concatenation in batch first, for the sake of faster convergence and better generalization. The overall feature vector is then fed into the four sublayers (256, 64, 16, and 1 hidden node, respectively) to complete the feature fusion. The sigmoid activation function is used to generate the prediction score of siRNA inhibition finally.
The architecture of our approach
To facilitate and streamline siRNA design, we construct an approach, named AttSiOff. Aside from the aforementioned siRNA inhibition predictor, it consists of an mRNA searching package and an off-target filter.
The mRNA searching package
Usually, probed mRNA sequences may update with the development of molecular biology, and only the target gene name is provided in the siRNA design. Downloading mRNA sequences manually on NCBI or other websites is time-consuming and annoying. Fortunately, we found one package, pyGB, implemented by Haotian Teng. For any given gene name, it could search for corresponding mature mRNAs rapidly.
The off-target filter
To build an effective siRNA design tool, we shall consider the off-target effects, which may weaken the siRNA inhibition, intervene in normal necessary cell activities, and do harm to receptors. They may arise from three aspects: on-target silencing of unintended mRNAs, miRNA-like offtarget silencing, and stimulation of innate immune response [28,29].
On-target unintended silencing results from 16 or more consecutive base pairings between the sequences of AS and unwanted mRNAs. It can be detected via substring searching algorithm. However, some effective sites do not confirm perfect pairings, which may allow for wobble or mismatch [30]. To solve this problem, we use an improved Smith-Waterman algorithm to calculate multiple optimal alignments between AS and unwanted mRNAs, by replacing the one-off backtracking with recurrent backtracking, until the current maximum score is less than a specific threshold.
MicroRNA-like off-target effect refers to siRNA-induced regulation of unintended transcripts, through partial sequence complementarity to their 3'UTRs [28]. The possible binding sites for miRNA are the 8mer site (base pairing at positions 2-8 with a base A opposite at position 1), 7merm8 site (base pairing at positions 2-8), and 7mer-A1 site (base pairing at positions 2-7 with a base A opposite position 1) (Supplementary Fig. S1) [30]. We can also search these base pairings by substring searching.
As for the non-specific immune response caused by siRNA, it can be reduced by selecting siRNAs carefully to avoid containing putative immunostimulatory motifs UGUGU and GUC CUU CAA in the AS [31]. Thus, we check if each siRNA contains the two motifs by substring searching.
The flow of our approach
The architecture of our siRNA design approach is shown in Fig. 2. The entire workflow is divided into four parts.

Fig. 2 The architecture of our siRNA design approach. It contains optional mRNA searching package pyGB, our siRNA inhibition prediction model, and optional off-target filter. The package pyGB can search mRNA sequences for given gene name from Genomes database. But one can also provide mRNA sequences directly. Substring and imprvoed Smith-Waterman algorithm are used to analyze on-target or miRNA-like offtarget effect, by aligning siRNA with each untargeted mRNA from Refseq or TargetScan 3'UTRs database |
First, the pyGB package is used to search for mature mRNAs according to the input gene names from the GENOMES database. One can also provide mRNA sequences in FASTA format directly.
Second, every mRNA sequence is cleaved to generate all alternative 19-nucleotide SSs via sliding window and get corresponding 21-nucleotide ASs, 59-nucleotide local target mRNA, and other attributes simultaneously. Considering the potential weak specificity and possible toxicity, the following characteristics in the AS are also collected in this step: the Gibbs free energy, presence of two immunostimulatory motifs, presence of long stretches of identical bases, GC content, GC content, presence of more than 2 continuous CAN pattern, and presence of more than 2 consecutive CUG/CCG/CGG motifs [32,33].
Third, every siRNA duplex and opposite local mRNAs are fed into our siRNA inhibition predictor, and all siRNAs targeting identical genes are sorted in prediction-descending order.
Fourth, one can choose an optional off-target filter for top-k siRNAs. Here, we mainly make use of two databases: humanRefseq (downloaded from ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/mRNA_Prot/human.rna.gbff.gz) and TargetScan (downloaded from https://www.targetscan.org/cgi-bin/targetscan/data_download.vert72.cgi). The human RefSeq database contains massive human mRNA sequences, and it is used to calculate on-target unintended silencing effects. The TargetScan database is composed of all human 3'UTR segments, and it is used to compute miRNA-like off-target effects. As is discussed above, the miRNA-like off-target and on-target off-target can both be calculated using substring searching. The on-target off-target can also be predicted by an improved Smith- Waterman algorithm.
This workflow will eventually provide the user with top-k siRNAs with their multiple attributes and possible off-target effects, which facilitate the pre-screening stage greatly.
Experimental setup
To ensure the uniform distribution between the trainset and the test set, we sample the siRNA with indices of i, i + 10, i + 20…in DHRT to form the test set during tenfold crossvalidation. Besides, we consider different sources of datasets as independent test set, to further validate the generalization of our method.
Our method is built with Pytorch in Python. We choose the Adam optimizer with weight decay of 5e−4 and an initial learning rate of 0.005. Dropout operation exists both in the multi-head self-attention module and the fully connected module, to repress the impact of overfitting. In addition, we set the maximum epoch to 1000. We use an early-stopping strategy to supervise the PCC metric. If this indicator continues decreasing for 20 epochs, it will terminate the training phase, and the model parameters with the best PCC will be saved. As a regression task, we train the model with the mean square error (MSE) loss.
Evaluation metrics
To estimate the prediction performance, we use three statistical indicators here, including the Pearson correlation coefficient (PCC), the Spearman correlation coefficient (SPCC), and the Area under the Receiver Operating Characteristic curve (AUC). PCC and SPCC evaluate the linear correlation between two sets of data. AUC is used to evaluate the performance of binary classification. In this paper, we use 0.7 as the inhibition threshold to classify a siRNA to be positive or negative.
Among these three metrics, SPCC is the most important one. It denotes the correlation of rankings between predictions and labels. In siRNA design, precise ranking prediction will reduce the workload to find functional siRNAs greatly. For example, a siRNA with a low inhibition of 0.2 is still optimal, if only its inhibition rank first.
Results and discussion
Tenfold cross-validation result
We compare our model with i-Score [11], Biopredsi [9], DSIR [10], one CNN-based model [12], and one GNNbased model [14]. The first three algorithms lack source code, and they are hard to reproduce. Fortunately, we find the i-Score webserver can generate all siRNA candidates with predictions of these models, for any given target mRNA sequence. But siRNAs at the first two positions are limited to predict, and that is why we discard two samples in DT above.
The tenfold cross-validation result is shown in Fig. 3. Apparently, our model achieves state-of-the-art performance among the six methods, reaching an average PCC of 0.81, SPCC of 0.84, and AUC of 0.886.

Fig. 3 The tenfold cross-validation result of our predictor compared with existing methods. Three metrics are shown with boxplots, including (a) PCC, (b) SPCC, and (c) AUC. The boxplot is used to describe 4-quartiles of data distribution. The orange lines represent mean values, the green dashed lines represent median values, and small circles represent outliers. Obviously, our method outperforms all existing methods among three metrics, reaching an average PCC of 0.81, SPCC of 0.84, and AUC of 0.886 |
In comparison, the three traditional methods show poor performance on all indicators. The reasons may be that their inputs, based on manual feature engineering, lack significant information and are usually biased. And their models have limited predictive capability to capture hidden motifs.
The CNN-based model reaches an average PCC of 0.67, SPCC of 0.652, and AUC of 0.848, which are much lower than ours. We may deduce that their convolutions with multiple kernels only focus on local adjacent correlation, and fail to capture the global interrelationship of the entire sequences. The forceful pooling operation results in the loss of significant information. Most importantly, their one-hot encoding representations suffer from the aforementioned problems, the sequence features obtained from which contain scanty information.
The performance of the GNN-based model is not bad, achieving an average PCC of 0.728, SPCC of 0.705, and AUC of 0.873. The idea of modeling siRNA-mRNA interaction tasks with graph structure is innovative and intuitive. Their experiments demonstrate that the biological graph model has potent predictive capability, even with limited features as the property of nodes. Their predictive accuracy may further improve with more meaningful features as input. However, the most serious problem of the GNN-based model is that it cannot predict on new siRNA sample with a fixed graph. That is why we exclude this model in the following cross-dataset validation.
Cross-dataset validation
For better demonstration of the generalization, we evaluate the cross-dataset performance. We train the model only on DH, DR, and test on DT, or train it only on DH, DT, and test on DR. These three datasets are independent of each other. There are two reasons why we do not consider DH as test set. One is that the samples in DH make up nearly seven-tenth of all samples, and it is very hard to train our predictor with a small amount of data. The other is that the three traditional models are all trained with DH. And we cannot get access to their source code or reproduce them easily. Instead, we collect their predictions on DT and DR from i-Score website. Therefore, the performance of them are unconvincing with DH as test set.
The results are shown in Table 2. It can be expected to notice a big drop in all metrics compared with those of tenfold cross-validation, due to the out-of-distribution problem.
Table 2 The cross-dataset prediction results. Bold numbers indicate the best results. Our method outperforms others on DT, while shows slight advantage on DR |
 |
Although the number of parameters in our model is much bigger than existing methods, it does not show the problem of overfitting. And it still significantly outperforms all other methods on cross-dataset validation, beyond our expectations. We may deduce that the batch normalization, dropout, and early-stopping strategy all help to ensure a good generalization.
To interpret the internal reasons why all indicators on DT are higher than those on DR, we use T-distributed Stochastic Neighbor Embedding (t-SNE) to visualize the RNA-FM embedding of siRNA sequences from three datasets (Supplementary Fig. S2). The samples in DT are intrinsically divided into three clusters, while the samples in DR are distributed more chaotically. That means predicting on DT is easier than that on DR coherently. We also analyze the differences in the inhibition distribution between these three datasets (Supplementary Fig. S3). The kernel density estimate (KDE) plots show that the distributions of DH and DT are quite similar, but they differ from DR. This may be another reason why all models perform better on DT than DR.
Comparison of five siRNA drugs
Moreover, we evaluate our method on above five siRNA drugs. They are chemically modified to further improve inhibition, lower off-target effect, and weaken toxicity. Theoretically, bare sequences of these drugs probably also show high inhibitions. If not, they should not be considered as candidates to add chemical modifications. Here we use their bare sequences to verify our method. The reported median knockdowns, the predicted inhibitions, the rankings of predictions among all corresponding siRNA candidates, and the number of possible off-target binding sites are shown in Supplementary Table S3.
Our approach, AttSiOff, helps a lot to facilitate this experiment. First, we take the target gene names as the input, namely TTR, ALAS1, PCSK9, and HAO1. The mRNA searching package then automatically searches the latest mature mRNA sequences from the GENOMES database and collects all possible siRNA candidates by sliding window. We do not need to download those mRNA sequences manually. Second, our predictor predicts the inhibitions of siRNAs grouped by gene names and sorts them in inhibitiondescending order. We use the location of these five siRNAs in their respective candidate sets as the ranking score. Third, for the five siRNAs, the off-target filter will compute the number of possible off-target binding sites.
The rankings show that inhibitions of the five siRNAs, predicted by our model, rank near the top in all candidates (1.79%, 0.62%, 2.4%, 11.2%, and 3.58%), which means researchers need fewer experiments to find wanted drugs, compared with other methods. And the predicted off-target effects are in allowable range, to ensure the specificity. The comparison results demonstrate that our predictor outperforms other methods and can facilitate the design process for a given gene.
The comparison results demonstrate that our predictor outperforms other methods and can facilitate the design process for a given gene.
Experiments on new target gene
We randomly select 40 siRNAs targeting AGT, to conduce biological experiments. However, we find that our method produces unsatisfactory result, reaching PCC of -0.268, SPCC of -0.231, and AUC of 0.441. It is quite difficult to work in unseen scenarios.
To solve this problem, we collect other 600 siRNAs targeting AGT from one patent (ID: WO2023014765A1). And these samples do not overlap with ours. After finetuning our pre-trained model, all indicators show great improvement, with PCC of 0.289, SPCC of 0.293, and AUC of 0.695. Focusing on those siRNAs with high experimental inhibitions, our finetuned model also gives high predictions. That means we would not miss potentially functional siRNAs.
Ablation study
More experiments are executed on the hyperparameters of TransformerEncoder. 2, 4, 8 layers, and 1, 2, and 4 heads are tested, separately. The results show that inadequate or excessive layers or heads both decrease the prediction performance. To obtain the optimal PCC metric, we select 4 layers with 4 heads for siRNA embedding and 2 layers with 1 head for mRNA embedding.
We also test the effect of different loss functions. In regression tasks, MAE and MSE are frequently used. The observed PCC is 0.712 for MAE and 0.77 for MSE. The reason may be that MSE loss will pay more attention to those anomalous samples to get more stable closed-loop solutions for weights. And this will lead to better generalization on the test set.
In addition, we emphasize the advantages of pre-trained RNA-FM embedding, compared with one-hot encoding. Thus, we also execute comparative experiments by replacing the RNA-FM embedding with one-hot encoding in a sequence context. The result gives a 21% average decrease in PCC.
Conclusions
Hundreds or thousands of siRNAs may target the same mRNA sequence. Utilizing computational methods to identify those hyper-functional siRNAs from massive candidates has increasingly become a significant study. However, existing methods are still not accurate and robust enough to design potent and specific siRNAs, and most of them have not considered unintended off-target effects.
In this paper, we propose a novel self-attention-based approach to siRNA inhibition and off-target effect prediction, named AttSiOff. First, the mRNA searching package helps obtain target mRNA sequences for a given gene name. Then the inhibition-related hidden features are captured from pre-trained RNA-FM embedding through the multi-head self-attention mechanism. After being concatenated with other prior knowledge-based features, they are fed into the fully connected module to complete the feature fusion and give the inhibition prediction. At last, the off-target filter utilizes substring searching or improved Smith-Waterman algorithms to give the possible amount of off-target binding sites. Compared with existing methods, our approach shows four major advantages. First, we include prior knowledge-based features as input, such as GC content, the secondary structure, etc. These features analyzed from a small biased dataset still help predict inhibitions more precisely. Second, we use a pre-trained RNA-FM model to encode the sequence context. The high-dimensional embedding contains more meaningful information than one-hot binary encoding, especially the functional and structural information. Third, we replace convolution with a multi-head self-attention mechanism, to capture the global long-distance dependencies within the entire sequence. Fourth, we use an additional filter to predict the off-target effects, to further ensure its specificity in practical application.
To evaluate the validity of our predictor, relevant comparison experiments are designed for verification. Experimental results show that our model achieves state-of-theart performance on PCC, SPCC, and AUC, compared with classical methods based on ANN, LASSO, SVM, CNN, and GNN. And the cross-dataset validation demonstrates the brilliant generalization and robustness of our model. Besides, our automatic siRNA design tool, AttSiOff, facilitates our experiments on five siRNA drugs, and we hope it can help other researchers, who are devoted to designing both effective and specific siRNA. In unseen scenarios, the predicator may produce unsatisfactory result. But it can be improved a lot, as long as there are some known samples to finetune pre-trained model. And this is perfectly normal for deep-learning-based algorithms.
This study provides new perspectives and analytical ideas for siRNA inhibition and off-target effects prediction. And we hope it helps bring self-attention mechanism to broader bioinformatics-related applications. Since some chemically modified siRNAs are deliberately designed to further improve their inhibitions and reduce the off-target effect, AttSiOff will be used as the backbone and the representations of chemical modification will be taken into account in the future.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s44258-024-00019-1.
Acknowledgements Not applicable.
Authors' contributions Conceptualization and original draft preparation, Bin Liu; reviewing and editing, Bin Liu, Ye Yuan; Ye Yuan, Xiaoyong Pan, Hong-Bin Shen, and Cheng Jin revised and provided critical feedback on the manuscript. All authors have read and agreed to the final version of the manuscript.
Funding This work was supported by grants from the National Natural Science Foundation of China (No. 62103262) and the Shanghai Pujiang Programme (No. 21PJ1407700)
Availability of data and materials Data and code are available at: https://github.com/2333liubin/AttSiOff
Declarations
Competing interests The authors declare no conflict of interest.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Publisher's Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}