内科理论与实践 ›› 2023, Vol. 18 ›› Issue (05): 328-333.doi: 10.16138/j.1673-6087.2023.05.004
周礼扬a, 张春丽b, 丁秋兰a, 李娅b( )
)
收稿日期:2023-03-09
出版日期:2023-10-30
发布日期:2024-01-25
通讯作者:
李娅
E-mail:yayayaly@126.com
基金资助:
ZHOU Liyanga, ZHANG Chunlib, DING Qiulana, LI Yab()
Received:2023-03-09
Online:2023-10-30
Published:2024-01-25
Contact:
LI Ya
E-mail:yayayaly@126.com
摘要: 目的 分析一个家族性多囊肾伴纤维蛋白原缺陷症家系的基因诊断及相关临床表现,研究其发病机制并为临床治疗提供指导。方法 将患者外周血样本中分离出基因组DNA,进行全外显子组测序,Sanger法对多囊肾病1(polycystic kidney disease 1,PKD1)和纤维蛋白原β链(fibrinogen beta chain,FGB)基因突变结果进行验证。肾脏超声检查多囊肾,Clauss法检测血浆纤维蛋白原活性,免疫比浊法检测血浆纤维蛋白原抗原。检索国内外2种疾病相关的文献并进行分析总结。结果 PKD1在第15外显子发生c.6586C>T,p.Q2196X杂合无义突变,FGB在第2外显子发生 c.130C>T,p.R44C杂合错义突变。患者超声结果诊断为多囊肾,纤维蛋白原抗原正常(2.3 g/L)、活性降低(1.25 g/L)。文献检索结果显示2种突变在国外均有报道,在国内首次报道。结论 PKD1 p.Q2196X和FGB p.R44C杂合突变分别导致该患者的多囊肾和异常纤维蛋白原血症。目前临床表型主要由多囊肾引起,应以多囊肾治疗为主,定期随访,保护肾功能。
中图分类号:
周礼扬, 张春丽, 丁秋兰, 李娅. 一个家族性多囊肾伴纤维蛋白原缺陷症家系的基因诊断、临床特征及文献回顾[J]. 内科理论与实践, 2023, 18(05): 328-333.
ZHOU Liyang, ZHANG Chunli, DING Qiulan, LI Ya. Genetic diagnosis and clinical analysis of congenital dysfibrinogenemia with polycystic disease: a case report and literature review[J]. Journal of Internal Medicine Concepts & Practice, 2023, 18(05): 328-333.
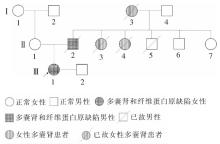
图1
多囊肾伴纤维蛋白原缺陷症家系
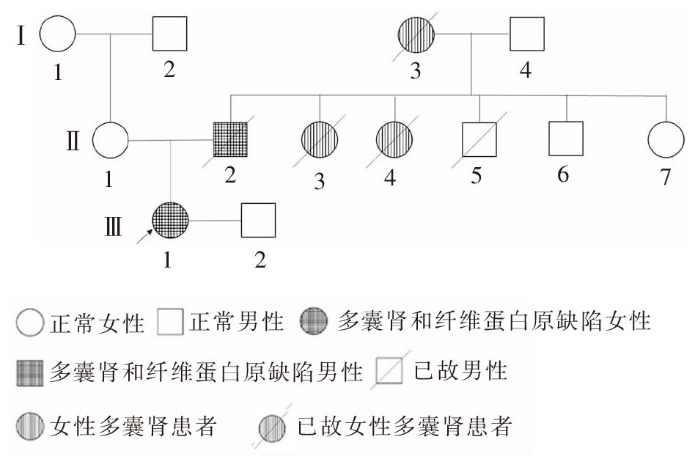
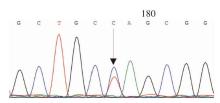
图2
PKD1第15外显子c.6586C>T,p.Q2196X
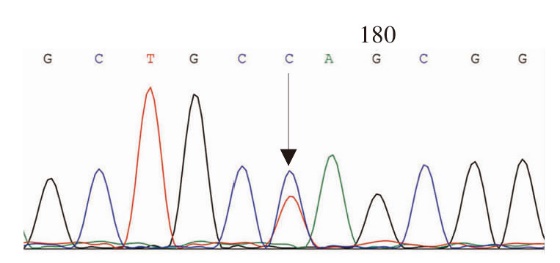
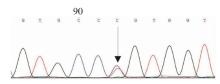
图3
FGB第2外显子c.130C>T,p.R44C
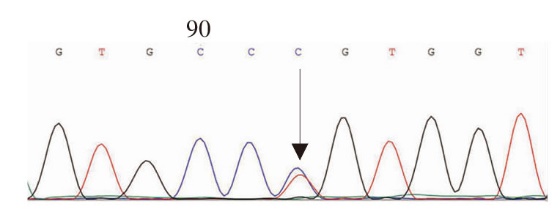
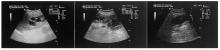
图4
患者腹部超声图
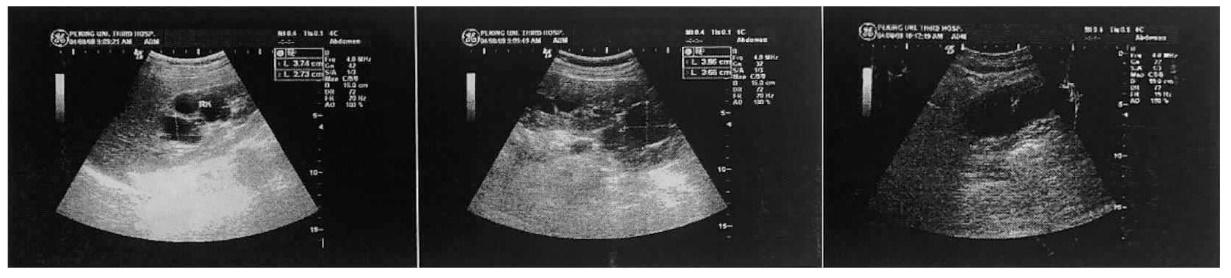
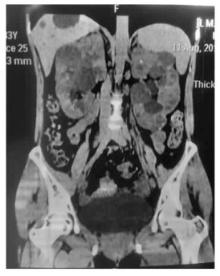
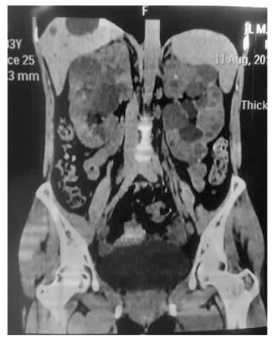
图5
患者腹部CT
| [1] |
Dell KM, Matheson M, Hartung EA, et al. Kidney disease progression in autosomal recessive polycystic kidney disease[J]. J Pediatr, 2016, 171: 196-201.
doi: 10.1016/j.jpeds.2015.12.079 URL |
| [2] |
Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease[J]. Nat Rev Dis Primers, 2018, 4(1):50.
doi: 10.1038/s41572-018-0047-y pmid: 30523303 |
| [3] | Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant[DB/OL]. 2022. https://www.ncbi.nlm.nih.gov/books/NBK1246/. |
| [4] |
Lanktree MB, Chapman AB. New treatment paradigms for ADPKD: moving towards precision medicine[J]. Nat Rev Nephrol, 2017, 13(12): 750-768.
doi: 10.1038/nrneph.2017.127 pmid: 28989174 |
| [5] |
Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease[J]. Lancet, 2019, 393(10174): 919-935.
doi: S0140-6736(18)32782-X pmid: 30819518 |
| [6] |
Neerman-Arbez M, de Moerloose P, Casini A. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders[J]. Semin Thromb Hemost, 2016, 42(4): 356-365.
doi: 10.1055/s-0036-1571340 pmid: 27019463 |
| [7] |
Richard M, Celeny D, Neerman-Arbez M. Mutations accounting for congenital fibrinogen disorders[J]. Semin Thromb Hemost, 2022, 48(8): 889-903.
doi: 10.1055/s-0041-1742170 URL |
| [8] |
Casini A, Neerman-Arbez M, Ariëns RA, et al. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management[J]. J Thromb Haemost, 2015, 13(6): 909-919.
doi: 10.1111/jth.12916 pmid: 25816717 |
| [9] |
Casini A, Blondon M, Lebreton A, et al. Natural history of patients with congenital dysfibrinogenemia[J]. Blood, 2015, 125(3): 553-561.
doi: 10.1182/blood-2014-06-582866 pmid: 25320241 |
| [10] |
Mangolini A, de Stephanis L, Aguiari G. Role of calcium in polycystic kidney disease[J]. World J Nephrol, 2016, 5(1): 76-83.
doi: 10.5527/wjn.v5.i1.76 URL |
| [11] |
Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease[J]. J Clin Invest, 2014, 124(6): 2315-2324.
doi: 10.1172/JCI72272 pmid: 24892705 |
| [12] |
Bergmann C. Recent advances in the molecular diagnosis of polycystic kidney disease[J]. Expert Rev Mol Diagn, 2017, 17(12): 1037-1054.
doi: 10.1080/14737159.2017.1386099 pmid: 28952822 |
| [13] |
Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD)[J]. Kidney Int, 2015, 88(1): 17-27.
doi: 10.1038/ki.2015.59 pmid: 25786098 |
| [14] |
Obeidova L, Elisakova V, Stekrova J, et al. Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease[J]. BMC Med Genet, 2014, 15: 41.
doi: 10.1186/1471-2350-15-41 pmid: 24694054 |
| [15] | Trujillano D, Bullich G, Ossowski S, et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing[J]. Mol Genet Genomic, 2014, 2(5): 412-421. |
| [16] |
Rahbari-Oskoui F, Williams O, Chapman A. Mechanisms and management of hypertension in autosomal dominant polycystic kidney disease[J]. Nephrol Dial Transplant, 2014, 29(12): 2194-2201.
doi: 10.1093/ndt/gft513 URL |
| [17] | Nauli SM, Jin X, Hierck BP. The mechanosensory role of primary cilia in vascular hypertension[J]. Int J Vasc Med, 2011: 376281. |
| [18] |
Kocyigit I, Eroglu E, Gungor O. Clinical problems in hemodialysis patients with autosomal dominant polycystic kidney disease[J]. Semin Dial, 2018, 31(3): 268-277.
doi: 10.1111/sdi.2018.31.issue-3 URL |
| [19] |
Rozenfeld MN, Ansari SA, Shaibani A, et al. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms?[J]. AJNR Am J Neuroradiol, 2014, 35(1): 3-9.
doi: 10.3174/ajnr.A3437 URL |
| [20] |
Perrone RD, Malek AM, Watnick T. Vascular complications in autosomal dominant polycystic kidney disease[J]. Nat Rev Nephrol, 2015, 11(10): 589-598.
doi: 10.1038/nrneph.2015.128 pmid: 26260542 |
| [21] |
Etminan N, Rinkel GJ. Unruptured intracranial aneurysms: development, rupture and preventive management[J]. Nat Rev Neurol, 2016, 12(12): 699-713.
doi: 10.1038/nrneurol.2016.150 pmid: 27808265 |
| [22] |
Cagnazzo F, Gambacciani C, Morganti R, et al. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management[J]. Acta Neurochir (Wien), 2017, 159(5): 811-821.
doi: 10.1007/s00701-017-3142-z URL |
| [23] |
Qian Q, Younge BR, Torres VE. Retinal arterial and venous occlusions in patients with ADPKD[J]. Nephrol Dial Transplant, 2007, 22(6): 1769-1771.
doi: 10.1093/ndt/gfm034 URL |
| [24] | Kuo IY, Chapman AB. Polycystins, ADPKD, and cardiovascular disease[J]. Kidney Int Rep, 2019, 5(4): 396-406. |
| [25] |
Gabow PA, Duley I, Johnson AM. Clinical profiles of gross hematuria in autosomal dominant polycystic kidney disease[J]. Am J Kidney Dis, 1992, 20(2): 140-143.
doi: 10.1016/s0272-6386(12)80541-5 pmid: 1496966 |
| [26] |
Colbert GB, Elrggal ME, Gaur L, et al. Update and review of adult polycystic kidney disease[J]. Dis Mon, 2020, 66(5): 100887.
doi: 10.1016/j.disamonth.2019.100887 URL |
| [27] |
Nishiura JL, Neves RF, Eloi SR, et al. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients[J]. Clin J Am Soc Nephrol, 2009, 4(4): 838-844.
doi: 10.2215/CJN.03100608 URL |
| [28] |
Torres VE, Wilson DM, Hattery RR, et al. Renal stone disease in autosomal dominant polycystic kidney disease[J]. Am J Kidney Dis, 1993, 22(4): 513-519.
pmid: 8213789 |
| [29] |
Gambaro G, Fabris A, Puliatta D, et al. Lithiasis in cystic kidney disease and malformations of the urinary tract[J]. Urol Res, 2006, 34(2): 102-107.
doi: 10.1007/s00240-005-0019-z pmid: 16416113 |
| [30] |
Moist LM, Churchill DN, House AA, et al. Regular monitoring of access flow compared with monitoring of venous pressure fails to improve graft survival[J]. J Am Soc Nephrol, 2003, 14(10): 2645-2653.
pmid: 14514744 |
| [31] |
Schwab SJ, Oliver MJ, Suhocki P, et al. Hemodialysis arteriovenous access: detection of stenosis and response to treatment by vascular access blood flow[J]. Kidney Int, 2001, 59(1): 358-362.
pmid: 11135091 |
| [32] |
Hsieh MY, Cheng CH, Chen CH, et al. The association of long-term blood pressure variability with hemodialysis access thrombosis[J]. Front Cardiovasc Med, 2022, 9:881454.
doi: 10.3389/fcvm.2022.881454 URL |
| [33] |
Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia[J]. Thromb Haemost, 1995, 73(1): 151-161.
doi: 10.1055/s-0038-1653741 URL |
| [34] | Casini A, How I treat dysfibrinogenemia[J]. Blood, 2021, 138(21): 2021-2030. |
| [35] |
Vakalopoulou S, Mille-Baker B, Mumford A, et al. Fibrinogen Bbeta14 Arg—>Cys: further evidence for a role in thrombosis[J]. Blood Coagul Fibrinolysis, 1999, 10(7): 403-408.
doi: 10.1097/00001721-199910000-00001 URL |
| [1] | 赵一菲 邹运 陈辉 林晓曦.
先天性黑素细胞痣基因突变类型分析及临床意义
[J]. 组织工程与重建外科杂志, 2023, 19(3): 258-. |
| [2] | 蒋怡然, 王卫庆. 原发性醛固酮增多症的分子机制研究进展[J]. 内科理论与实践, 2023, 18(04): 261-265. |
| [3] | 韩钰钰 余明薇 王慜 徐媛 陈勇 袁斯明. 体表软组织静脉畸形中局限性血管内凝血的临床特征分析[J]. 组织工程与重建外科杂志, 2022, 18(3): 214-. |
| [4] | 杨崔燕, 王豪雨, 陈小松, 沈坤炜. 抑癌基因TP53突变状态与三阴性乳腺癌病人预后的研究[J]. 外科理论与实践, 2022, 27(05): 421-428. |
| [5] | 郝旭, 王伟铭. 依靠肾活检确诊的以肾脏病变为主要表现的法布里病1例报告[J]. 诊断学理论与实践, 2022, 21(04): 527-529. |
| [6] | 徐娜娜, 吴涛, 寇明坤, 白海. ASXL1基因突变在急性髓系白血病中的研究进展[J]. 内科理论与实践, 2022, 17(04): 353-355. |
| [7] | 蔡晓婷, 易华华, 林佳媛, 陈聆. 常染色体显性多囊肾合并肺栓塞一例并文献复习[J]. 诊断学理论与实践, 2022, 21(01): 80-85. |
| [8] | 周璐, 雷航, 洪叶, 金爽, 董永勤, 王学锋, 蔡晓红. 一个新的ABO*A等位基因导致的AwB亚型及其分子机制研究[J]. 诊断学理论与实践, 2021, 20(06): 547-551. |
| [9] | 姚碧莲, 张欣欣, 韩悦. 肝豆状核变性的基因诊断研究进展[J]. 内科理论与实践, 2021, 16(05): 354-358. |
| [10] | 赵英妹, 聂红明, 张珏, 黄燕. 肝硬化患者凝血五项的测定及其临床意义[J]. 诊断学理论与实践, 2021, 20(05): 480-483. |
| [11] | 崔恒庆,孙滨,方霞,周晟博,杨皓然,戴心怡,韩刚,王斌. NOG R167G突变致先天性指间关节黏连[J]. 组织工程与重建外科杂志, 2020, 16(1): 39-42. |
| [12] | 雷航, 范亮峰, 蔡晓红, 王钰箐, 刘曦, 金沙, 沈伟, 陆琼, 向东, 王学锋, 邹纬. 中国人群血型ABO亚型的分子基础研究[J]. 诊断学理论与实践, 2020, 19(04): 364-369. |
| [13] | 姜逊渭, 王健怡, 肖婷婷, 谢利剑, 侯翠兰, 张永为,. DSP复合杂合基因突变与扩张型心肌病的发生有关[J]. 内科理论与实践, 2020, 15(02): 94-98. |
| [14] | 彭真萍, 项喜喜, 张苏江, 李佳明. 以类白血病反应为首发表现的慢性中性粒细胞白血病二例并文献复习[J]. 诊断学理论与实践, 2020, 19(02): 122-128. |
| [15] | 彭美芳,吴颖之,陈洁仪,金力,穆雄铮,汪思佳. FGFR2突变位点Ser252Trp与Pro253Arg对Apert综合征临床表型影响的差异—基于荟萃分析的证据[J]. 组织工程与重建外科杂志, 2018, 14(2): 67-72. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||